TOBLAPTIN®
GADOR
Trat.Poliquistosis Renal Autosómica Dominante.
Composición.
Cada comprimido de TOBLAPTIN® 15 contiene: Tolvaptan 15,000 mg. Excipientes: Hidroxipropilcelulosa EXF 9,513 mg, Lactosa monohidrato 34,638 mg, Almidón de maíz 10,000 mg, Celulosa microcristalina PH101 10,000 mg, Indigotina 0,004 mg, Hidroxipropilcelulosa LH22 4,782 mg, Estearato de magnesio vegetal 1,063 mg. Cada comprimido de TOBLAPTIN® 30 contiene: Tolvaptan 30,000 mg. Excipientes: Hidroxipropilcelulosa EXF 19,026 mg, Lactosa monohidrato 69,276 mg, Almidón de maíz 20,000 mg, Celulosa microcristalina PH101 20,000 mg, Indigotina 0,008 mg, Hidroxipropilcelulosa LH22 9,564 mg, Estearato de magnesio vegetal 2,126 mg.
Farmacología.
Mecanismo de acción: Tolvaptán es un antagonista de la vasopresina que bloquea específicamente la unión de la vasopresina arginina (AVP) a los receptores V2 en las porciones distales de la nefrona. La afinidad de tolvaptán por el receptor V2 humano es 1,8 veces superior a la de la AVP nativa. Tolvaptán antagoniza el efecto de la vasopresina y produce un aumento en la excreción urinaria de agua que resulta en un incremento en la depuración de agua libre (acuaresis), una osmolalidad de la orina y como consecuencia, la elevación de las concentraciones séricas de sodio. La excreción urinaria de sodio y potasio, y las concentraciones plasmáticas de potasio no cambian significativamente. Los metabolitos de tolvaptán, en comparación con tolvaptán, no tienen o tienen una actividad antagonista leve ante los receptores V2 humanos. Las concentraciones plasmáticas de AVP nativa pueden aumentar (promedio 2-9 pg/ml) con la administración de tolvaptán. Efectos farmacodinámicos Se han determinado los efectos farmacodinámicos de tolvaptán en sujetos sanos y en sujetos con PQRAD en estadio 1 a 4 de ERC. En sujetos adultos sanos, la administración oral de dosis de 7,5 a 120 mg de tolvaptán produjo un incremento de la tasa de excreción urinaria en un plazo de 2 horas desde la administración. Después de dosis orales únicas de 7,5 a 60 mg, el volumen de orina de 24 horas aumentó de forma dependiente de la dosis, con volúmenes diarios que variaron desde 3 hasta 9 litros. Para todas las dosis, las tasas de excreción de orina regresaron a los niveles basales después de 24 horas. Después de dosis únicas de 60 a 480 mg, se excretó una media de unos 7 litros durante 0 a 12 horas, con independencia de la dosis administrada. Dosis notablemente superiores de tolvaptán producen respuestas más mantenidas sin afectar a la magnitud de la excreción, ya que las concentraciones activas de tolvaptán están presentes durante períodos más largos de tiempo. Luego de la administración de una dosis única de 60 mg de tolvaptán, el aumento de los niveles de sodio ocurre entre las 2 a 4 horas posteriores a la dosis; entre 4 y 8 horas luego de la dosis, se observó un efecto máximo con un aumento de aproximadamente 6 mEq en el sodio sérico y un aumento de aproximadamente 9 ml/min en la velocidad de excreción urinaria. Por lo tanto, el efecto farmacológico presenta un retraso respecto de concentraciones plasmáticas de tolvaptán. Alrededor del 60% del efecto máximo en el sodio sérico se mantiene hasta 24 horas posteriores a la dosis, pero la tasa de excreción urinaria ya no se mantiene aumentada ese tiempo. Una dosis superior a 60 mg de tolvaptán no produce mayores incrementos de la acuaresis o el sodio sérico. Los efectos de tolvaptán en el rango de dosis recomendado de 15 a 60 mg una vez al día para el tratamiento de la hiponatremia, parece estar limitado a acuaresis y al aumento resultante en la concentración de sodio. Los efectos del aclaramiento de agua libre y del volumen de orina resultan evidentes en todos los estadios de la ERC, observándose efectos absolutos más reducidos en los estadios posteriores, lo que es coherente con la disminución del número de nefronas totalmente funcionales. Las reducciones agudas en el volumen renal total medio se observaron también al cabo de 3 semanas de tratamiento, en todos los estadios de ERC, desde -4,6 % en la ERC en estadio 1 hasta -1,9 % en la ERC en estadio 4. No se observaron efectos significativos en el intervalo QTc luego de la administración de 30 mg y 300 mg de tolvaptán en los días 1 y 5. En la dosis de 300 mg, se observó un pico plasmático de tolvaptán aproximadamente 4 veces mayor que la concentración máxima alcanzada luego de la dosis de 30 mg. Luego de la administración de moxifloxacina el intervalo QT aumentó en 12 ms a las 2 horas el día 1 y 17 ms después de 1 hora el día 5 luego del tratamiento, lo que indica que el estudio fue diseñado y llevado a cabo adecuadamente para detectar el efecto de tolvaptán en el intervalo QT, el cual estuvo presente.
Farmacocinética.
Absorción: Tras la administración oral, tolvaptán se absorbe rápidamente, observándose el pico de las concentraciones plasmáticas aproximadamente 2 horas después de la administración. La biodisponibilidad absoluta del tolvaptán es de aproximadamente el 56 %. La coadministración de tolvaptán junto con una comida con alto contenido en grasas aumentó las concentraciones máximas de tolvaptán hasta incluso duplicarlas, si bien no se observaron cambios en el ABC. Si bien se desconoce la relevancia clínica de este hallazgo, la dosis matutina se debe tomar en ayunas con el fin de reducir al mínimo el riesgo innecesario de aumentar la exposición máxima. Distribución: Tras dosis orales únicas de 300 mg, las concentraciones plasmáticas máximas parecen estabilizarse, posiblemente debido a la saturación de la absorción. Tolvaptán se fija de manera reversible (98 %) a las proteínas plasmáticas. Metabolismo: Tolvaptán es ampliamente metabolizado en el hígado, prácticamente en exclusiva por el CYP3A. Tolvaptán es un sustrato débil del CYP3A y no parece tener actividad inhibitoria. Los estudios in vitro indicaron que tolvaptán no tiene ninguna actividad inhibitoria del CYP3A. Se han identificado catorce metabolitos en el plasma, la orina y las heces; todos menos uno de ellos también fueron metabolizados por el CYP3A. El metabolito ácido oxobutírico es el único presente con una concentración mayor del 10 % de la radioactividad plasmática total; todos los demás metabolitos están presentes a concentraciones más bajas que el tolvaptán. Los metabolitos de tolvaptán contribuyen entre poco y nada al efecto farmacológico de tolvaptán; todos los metabolitos presentan una actividad antagonista entre nula y débil respecto a los receptores V2 humanos, en comparación con tolvaptán. La semivida de eliminación terminal es de aproximadamente 8 horas y se alcanza el estado estacionario de las concentraciones de tolvaptán tras la primera dosis. Eliminación: Menos del 1 % del principio activo intacto se excreta inalterado en la orina. Los experimentos con tolvaptán marcado radioactivamente mostraron que se recuperó el 40 % de la radioactividad en la orina y el 59 % en las heces, mientras que tolvaptán inalterado representó el 32 % de la radioactividad. Tolvaptán es únicamente un componente menor del plasma (3 %). Linealidad/no linealidad: Después de dosis orales únicas, los valores de la Cmáx resultan menores que los aumentos proporcionales de dosis de 30 mg a 240 mg y después presentan una concentración estable con dosis de entre 240 mg y 480 mg, la ABC aumenta linealmente. Tras la administración de varias dosis diarias de 300 mg, la exposición a tolvaptán solo aumentó 6,4 veces en comparación con una dosis de 30 mg. En el caso de los regímenes con dosis dividida de 30 mg/día, 60 mg/día y 120 mg/día en pacientes con poliquistosis renal autosómica dominante, la exposición (ABC) a tolvaptán aumenta linealmente. Poblaciones especiales: Insuficiencia hepática: Se ha investigado el efecto de una función hepática leve o moderadamente disminuida (clases A y B en la escala Child-Pugh) sobre la farmacocinética de tolvaptán en pacientes con hepatopatías de orígenes diversos. No se han observado cambios clínicamente significativos en el aclaramiento de las dosis entre 5 mg y 60 mg. Se dispone de información muy limitada sobre el caso de los pacientes con insuficiencia hepática grave (clase C en la escala Child-Pugh). En un análisis farmacocinético de la población en pacientes con insuficiencia hepática, las ABC de tolvaptán en pacientes con una insuficiencia hepática grave (clase C en la escala Child-Pugh) y leve o moderada (clases A y B en la escala Child-Pugh) fueron 3,1 veces y 2,3 veces más elevadas que en los sujetos sanos. En pacientes con hiponatremia de cualquier origen, la depuración de tolvaptán se reduce a aproximadamente 2 ml/min/kg. La insuficiencia hepática moderada o severa o la insuficiencia cardíaca disminuyen la depuración y aumentan el volumen de distribución de tolvaptán; sin embargo, dichos cambios no son clínicamente relevantes. Insuficiencia renal: En un análisis de farmacocinética poblacional de pacientes con insuficiencia cardíaca, las concentraciones de tolvaptán de pacientes con afectación de la función renal leve (aclaramiento de creatinina [Ccr] 50 a 80 ml/min) o moderada (Ccr 20 a 50 ml/min) no fueron significativamente distintas a las concentraciones de tolvaptán en pacientes con una función renal normal (Ccr 80 a 150 ml/min). El inicio y la compensación del efecto de tolvaptán en el sodio sérico fueron más lentos en pacientes con insuficiencia renal severa. En un análisis farmacocinético de la población de pacientes con poliquistosis renal autosómica dominante, las concentraciones de tolvaptán aumentaron, en comparación con los sujetos sanos, a medida que la función renal se situó por debajo de una TFGe de 60 ml/min/1,73 m2. Se asoció una reducción de TFGeERC-EPI de 72,2 a 9,79 (ml/min/1,73 m2) a una reducción del 32 % en el aclaramiento corporal total. No se han evaluado la eficacia y la seguridad de tolvaptán en quienes tienen un aclaramiento de creatinina < 10 ml/min y, por lo tanto, se desconocen. Edad: El aclaramiento de tolvaptán no se ve afectado significativamente por la edad.
Indicaciones.
Hiponatremia: TOBLAPTIN® está indicado para el tratamiento de la hiponatremia hipervolémica o euvolémica clínicamente significativa (sodio sérico menor a 125 mEq/l o hiponatremia menos marcada pero sintomática y resistente a la corrección por restricción de líquidos), incluyendo pacientes con insuficiencia cardíaca y síndrome de secreción inadecuada de hormona antidiurética (SIHAD). No debe tratarse con TOBLAPTIN® a aquellos pacientes que requieren intervención para aumentar el sodio sérico con urgencia para prevenir o tratar síntomas neurológicos graves. Poliquistosis renal autosómica dominante: TOBLAPTIN® está indicado para ralentizar la progresión del desarrollo de quistes y la insuficiencia renal asociada a poliquistosis renal autosómica dominante (PQRAD) en adultos con enfermedad renal crónica (ERC) en estadio 1 a 4 al inicio del tratamiento y con signos de enfermedad de progresión rápida.
Dosificación.
Hiponatremia: El tratamiento con TOBLAPTIN® se debe iniciar en el hospital debido a la necesidad de realizar una fase de ajuste de la dosis que requiere una estrecha monitorización del sodio sérico y del estado volémico. Posología: TOBLAPTIN® se debe iniciar a una dosis de 15 mg una vez al día. Luego de al menos 24 horas, aumentar la dosis a 30 mg. La dosis podría incrementarse hasta un máximo de 60 mg una vez al día según sea tolerada para conseguir el nivel deseado de sodio sérico. TOBLAPTIN® debe administrarse preferiblemente por las mañanas, con independencia de que se tome con o sin alimentos. TOBLAPTIN® no se debe tomar con jugo de pomelo. Dosis más bajas de tolvaptán podrían ser necesarias en pacientes con riesgo de corrección demasiado rápida del sodio (por ejemplo, en pacientes con enfermedades oncológicas, niveles basales de sodio sérico muy bajos, que toman diuréticos o que toman suplementos de sodio). En estos casos, las formulaciones de TOBLAPTIN® 15 mg y 30 mg podrían no ser adecuadas. Durante el ajuste de la dosis, se debe monitorizar tanto el sodio sérico como el estado volémico de los pacientes. Se debe evitar la restricción de líquidos durante las primeras 24 horas de tratamiento. Se debe informar a los pacientes tratados con TOBLAPTIN® que pueden tomar líquido si tienen sed. En caso de que no se produzca un incremento adecuado de los niveles de sodio sérico, se deben considerar otras opciones de tratamiento, tanto en lugar de tolvaptán o como tratamiento coadyuvante a éste. El uso de tolvaptán en combinación con otras opciones puede aumentar el riesgo de una corrección demasiado rápida del sodio sérico. En aquellos pacientes en los que se consiga un incremento adecuado de los niveles de sodio séricos, deben monitorizarse a intervalos regulares la enfermedad subyacente y los niveles de sodio sérico, para evaluar si es necesario continuar el tratamiento con tolvaptán. En el contexto de la hiponatremia, la duración del tratamiento queda determinada por la enfermedad subyacente y su tratamiento. Se espera que el tratamiento con tolvaptán continúe hasta que la enfermedad subyacente haya sido tratada adecuadamente o hasta el momento en que la hiponatremia haya dejado de ser un problema clínico. Con el fin de minimizar el riesgo de daño hepático, TOBLAPTIN® no debe administrarse durante más de 30 días para el tratamiento de la hiponatremia. Luego de suspender el tratamiento con TOBLAPTIN®, se debe recomendar a los pacientes que reanuden la restricción de líquidos, y se deben controlar los cambios en el sodio sérico y el volumen sérico. Poliquistosis renal autosómica dominante: El tratamiento con TOBLAPTIN® se debe iniciar y continuar bajo la supervisión de un profesional médico con experiencia en el tratamiento de la PQRAD y que conozca completamente los riesgos del tratamiento con tolvaptán, incluida la toxicidad hepática y los requisitos de monitoreo. Posología: TOBLAPTIN® se debe administrar dos veces al día, con un régimen de dosis dividida de 45 mg (3 comprimidos de TOBLAPTIN® 15 mg) + 15 mg, 60 mg (2 comprimidos de TOBLAPTIN® 30 mg) + 30 mg o 90 mg (3 comprimidos de TOBLAPTIN® 30 mg) + 30 mg. La dosis matutina se deberá tomar al menos 30 minutos antes del desayuno. La segunda dosis diaria se podrá tomar con o sin alimentos. De acuerdo con estos regímenes de dosis dividida, las dosis totales diarias serán de 60 mg, 90 mg o 120 mg. Modificaciones de la dosis recomendada: La dosis inicial es de 60 mg de tolvaptán al día con un régimen de dosis dividida de 45 mg (3 comprimidos de TOBLAPTIN® 15 mg al levantarse, antes de desayunar) + 15 mg (8 horas después). La dosis inicial se puede ajustar al alza hasta un régimen de dosis dividida de 90 mg de tolvaptán [60 mg (2 comprimidos de TOBLAPTIN® 30 mg) + 30 mg] al día y, a partir de ahí, a un régimen de dosis dividida de 120 mg de tolvaptán [90 mg (3 comprimidos de TOBLAPTIN® 30 mg) + 30 mg] al día, si se toleran, con un intervalo de al menos una semana entre cada ajuste de dosis. La dosis se debe ajustar con cuidado, para asegurarse de que no haya una mala tolerancia a dosis altas como consecuencia de un ajuste al alza demasiado rápido. La dosis se podrá volver a ajustar a la baja dependiendo de la tolerabilidad. Los pacientes se deben mantener con la dosis más alta de TOBLAPTIN® que puedan tolerar. El objetivo del ajuste de dosis es bloquear la actividad de la vasopresina en el receptor renal V2 de la manera más completa y constante posible, a la vez que se mantiene un equilibrio hídrico aceptable. Se recomienda realizar mediciones de la osmolalidad urinaria para monitorizar si la inhibición de la vasopresina es adecuada. La monitorización periódica de la osmolalidad plasmática o de la concentración sérica de sodio (para calcular la osmolalidad plasmática) y/o el peso corporal se deben emplear para monitorizar el riesgo de deshidratación asociada a los efectos acuaréticos de tolvaptán en caso de que el paciente ingiera una cantidad insuficiente de agua. No se ha evaluado la seguridad y la eficacia de tolvaptán en pacientes con ERC en estadio 5. Por este motivo, se debe interrumpir el tratamiento con tolvaptán si la insuficiencia renal progresa a ERC en estadio 5. Se debe interrumpir el tratamiento cuando la capacidad para beber o el acceso al agua estén limitados. TOBLAPTIN® no se debe tomar con jugo de pomelo. Se debe indicar a los pacientes que beban una cantidad suficiente de agua u otros líquidos acuosos. Ajuste de la dosis en pacientes tratados con inhibidores potentes del CYP3A: En pacientes que tomen inhibidores potentes del CYP3A, las dosis de tolvaptán se deben reducir de la siguiente manera: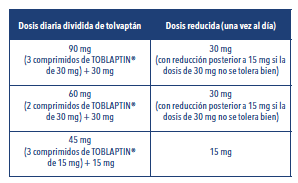
Ajuste de la dosis en pacientes tratados con inhibidores moderados del CYP3A En pacientes que tomen inhibidores moderados del CYP3A, las dosis de tolvaptán se deben reducir de la siguiente manera: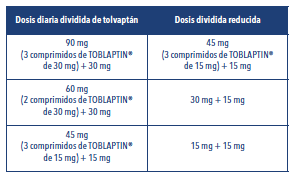
Se tienen que considerar nuevas reducciones de dosis si los pacientes no toleran bien las dosis reducidas de tolvaptán. Poblaciones especiales: Insuficiencia renal: Tolvaptán está contraindicado en pacientes anúricos. No se requiere un ajuste de la dosis en los pacientes con insuficiencia renal. No se han llevado a cabo ensayos clínicos con sujetos con índices de filtración glomerular < 10 ml/min ni con pacientes sometidos a diálisis. No se recomienda el luso de tolvaptán en pacientes con índices de filtración glomerular < 10 ml/min. El riesgo de daño hepático en pacientes con una función renal gravemente deteriorada (esto es, tasa de filtración glomerular estimada [TFGe] < 20) puede ser más elevado; estos pacientes deben ser supervisados estrechamente para detectar una posible hepatotoxicidad. Los datos sobre pacientes con ERC en estadio 4 temprano son más limitados que los de los pacientes en estadio 1, 2 o 3. No hay datos disponibles para los pacientes con ERC en estadio 4 tardío (TFGe < 25 ml/min/1,73 m2) y estadio 5. Se debe interrumpir el tratamiento con tolvaptán si la insuficiencia renal progresa a ERC en estadio 5. Insuficiencia hepática: En pacientes con insuficiencia hepática grave, se deben valorar detenidamente los riesgos y beneficios del tratamiento con TOBLAPTIN®. Los pacientes deben ser tratados con cuidado y se deben monitorizar regularmente las enzimas hepáticas. TOBLAPTIN® está contraindicado en pacientes con valores elevados de las enzimas hepáticas y/o signos o síntomas de daño hepático antes del inicio del tratamiento, y que cumplan los requisitos para la interrupción permanente del tratamiento con tolvaptán. No es necesario ajustar la dosis en pacientes con insuficiencia hepática leve o moderada (clases A y B en la escala Child-Pugh). Población de edad avanzada: El aumento de la edad no tiene ningún efecto sobre las concentraciones plasmáticas de tolvaptán. Se dispone de datos limitados sobre la seguridad y la eficacia de tolvaptán en pacientes con PQRAD de más de 55 años de edad. Población pediátrica: No se ha establecido todavía la seguridad y eficacia de tolvaptán en niños y adolescentes. No se dispone de datos. No se recomienda el uso de TOBLAPTIN® en pacientes pediátricos. Modo de administración: TOBLAPTIN® se administra por vía oral. Los comprimidos se deben tragar sin masticar y acompañados de un vaso de agua. TOBLAPTIN® no se debe tomar con jugo de pomelo.
Contraindicaciones.
Hipersensibilidad al principio activo o a alguno de los excipientes, o a la benzazepina o derivados de la benzazepina. Concentración elevada de enzimas hepáticas y/o signos o síntomas de daño hepático antes del inicio del tratamiento y cumplimiento de los requisitos para la interrupción permanente del tratamiento con tolvaptán. Anuria, ya que no se espera un beneficio clínico en pacientes que no pueden orinar. Hipovolemia, dado que los riesgos asociados al empeoramiento de la hipovolemia, incluidas las complicaciones como hipotensión e insuficiencia renal, superan los posibles beneficios. Hiponatremia hipovolémica. Hipernatremia. Pacientes incapaces de percibir o responder a la sensación de sed. Embarazo. Lactancia.
Reacciones adversas.
Reacciones adversas notificadas en el tratamiento de la hiponatremia Las reacciones adversas más frecuentes (incidencia mayor o igual al 5% en relación al placebo) observadas en estudios clínicos de hiponatremia doble ciego, controlados con placebo, de 30 días de tratamiento con tolvaptán a dosis ajustadas (15 mg a 60 mg una vez al día), fueron sed, sequedad de boca, astenia, constipación, polaquiuria o poliuria e hiperglucemia. A continuación se enumeran las reacciones adversas aparecidas durante el tratamiento con tolvaptán, agrupadas según el sistema de clasificación de órganos y por frecuencia. Las frecuencias se definen de la siguiente forma: frecuentes (mayor del 10 %); ocasionales (1 % al 10 %); raras (menor del 1 %). Las reacciones adversas se presentan en orden decreciente de gravedad dentro de cada intervalo de frecuencia. La frecuencia de las reacciones adversas notificadas durante el uso después de la comercialización no puede ser determinada, ya que se derivan de notificaciones espontáneas. Por lo tanto, la frecuencia de estas reacciones adversas se califica como "no conocida". Trastornos del sistema inmunológico. Frecuencia no conocida: reacciones de hipersensibilidad que incluyen shock anafiláctico, erupción generalizada. Trastornos de la sangre y del sistema linfático. Raras: coagulación intravascular diseminada. Trastornos del metabolismo y la nutrición. Ocasionales: polidipsia, deshidratación, hipercalcemia, hiperglucemia, hipoglucemia1, hipernatremia1, hiperuricemia1, apetito disminuido. Raras: cetoacidosis diabética. Trastornos del sistema nervioso. Ocasionales: síncope1, cefalea1, mareo. Raras: disgeusia, accidente cerebrovascular. Frecuencia no conocida: síndrome de desmielinización osmótica. Trastornos cardíacos. Raras: trombo intracardíaco, fibrilación ventricular. Trastornos vasculares. Ocasionales: hipotensión ortostática. Raras: trombosis venosa profunda. Trastornos respiratorios, torácicos y mediastínicos. Raras: embolia pulmonar, insuficiencia respiratoria. Trastornos gastrointestinales. Frecuentes: náuseas. Ocasionales: estreñimiento, diarrea1, xerostomía, sangrado gastrointestinal en pacientes con cirrosis. Raras: colitis isquémica. Trastornos hepatobiliares. Frecuencia no conocida: trastornos hepáticos2, insuficiencia hepática aguda3. Trastornos de la piel y del tejido subcutáneo. Ocasionales: equimosis, prurito. Raras: erupción pruriginosa1. Trastornos musculoesqueléticos y del tejido conjuntivo. Raras: rabdomiólisis. Trastornos renales y urinarios. Ocasionales: polaquiuria, poliuria. Raras: insuficiencia renal, hemorragia uretral. Trastornos del sistema reproductivo y de la mama. Raras: hemorragia vaginal. Trastornos generales y alteraciones en el lugar de administración. Frecuentes: sed. Ocasionales: astenia, pirexia, malestar1. Exploraciones complementarias. Ocasionales: presencia de sangre en orina1, alanina aminotransferasa elevada1, aspartato aminotransferasa elevada1, aumento de la creatinina en sangre. Raras: bilirrubina elevada1, tiempo de protrombina prolongado. Frecuencia no conocida: transaminasas elevadas2, hipernatremia. Procedimientos médicos y quirúrgicos. Frecuentes: corrección rápida de la hiponatremia, que en ocasiones da lugar a síntomas neurológicos. ¹ Observados en ensayos clínicos que investigan otras indicaciones. ² A partir de un estudio de seguridad posterior a la autorización sobre hiponatremia secundaria a SIHAD. ³ Observado en la supervisión posterior a la comercialización de tolvaptán en PQRAD; fue necesario un trasplante de hígado. Descripción de reacciones adversas seleccionadas: Corrección rápida de la hiponatremia: En un estudio de seguridad posterior a la autorización de tolvaptán sobre hiponatremia secundaria a SIHAD, que incluyó una alta proporción de pacientes con tumores (especialmente con cáncer de pulmón microcítico), de pacientes con niveles basales de sodio sérico bajos y de pacientes con uso concomitante de diuréticos y/o solución de cloruro de sodio, se mostró que la incidencia de la corrección rápida de la hiponatremia era más elevada que en los ensayos clínicos. Reacciones adversas notificadas en el tratamiento de la poliquistosis renal autosómica dominante: Las reacciones adversas predecibles desde el punto de vista farmacodinámico y notificadas con mayor frecuencia son sed, poliuria, nicturia y polaquiuria, que tuvieron lugar respectivamente en el 55 %, el 38 %, el 29 % y el 23 % de los pacientes. Además, tolvaptán se ha asociado a elevaciones idiosincrásicas de las concentraciones sanguíneas de alanina transaminasa (ALT; 4,4 %) y de aspartato transaminasa (AST; 3,1 %), con casos poco frecuentes de elevaciones concomitantes de la bilirrubina total (BT; 0,2 %). A continuación se enumeran las reacciones adversas aparecidas durante el tratamiento con tolvaptán, agrupadas según el sistema de clasificación de órganos y por frecuencia. Las frecuencias se definen de la siguiente forma: frecuentes (mayor del 10 %); ocasionales (1 % al 10 %); raras (menor del 1 %). Las reacciones adversas se presentan en orden decreciente de gravedad dentro de cada intervalo de frecuencia. La frecuencia de las reacciones adversas notificadas durante el uso después de la comercialización no puede ser determinada, ya que se derivan de notificaciones espontáneas. Por lo tanto, la frecuencia de estas reacciones adversas se califica como "no conocida". Trastornos del sistema inmunológico. Frecuencia no conocida: shock anafiláctico, erupción generalizada. Trastornos del metabolismo y la nutrición. Frecuentes: polidipsia. Ocasionales: deshidratación, hipernatremia, apetito disminuido, hiperuricemia, hiperglucemia, gota. Trastornos psiquiátricos. Ocasionales: insomnio. Trastornos del sistema nervioso. Frecuentes: cefalea, mareo. Trastornos cardíacos. Ocasionales: palpitaciones. Trastornos respiratorios, torácicos y mediastínicos. Ocasionales: disnea. Trastornos gastrointestinales. Frecuentes: diarrea, boca seca. Ocasionales: dolor abdominal, distensión abdominal, estreñimiento, dispepsia, enfermedad por reflujo gastroesofágico. Trastornos hepatobiliares. Ocasionales: función hepática anormal. Frecuencia no conocida: insuficiencia hepática aguda (observado en la supervisión posterior a la comercialización de tolvaptán en PQRAD; fue necesario un trasplante de hígado). Trastornos de la piel y del tejido subcutáneo. Ocasionales: erupción, prurito. Trastornos musculoesqueléticos y del tejido conjuntivo. Ocasionales: espasmos musculares. Trastornos renales y urinarios. Frecuentes: nicturia, polaquiuria, poliuria. Trastornos generales y alteraciones en el lugar de administración. Frecuentes: fatiga, sed. Ocasionales: astenia. Exploraciones complementarias. Ocasionales: alanina aminotransferasa elevada, aspartato aminotransferasa elevada, peso disminuido. Raras: bilirrubina elevada. Descripción de reacciones adversas seleccionadas: Resultados analíticos: Se observó una elevación ( > 3 × límite superior de la normalidad [LSN]) de la ALT en el 4,4 % de los pacientes tratados con tolvaptán y en el 1,0 % de los pacientes con placebo, al tiempo que se observó una elevación ( > 3 × LSN) de la AST en el 3,1 % de los pacientes tratados con tolvaptán y el 0,8 % de los pacientes con placebo en un ensayo con doble enmascaramiento, controlado por placebo en pacientes con PQRAD. Dos (0,2 %) de estos pacientes tratados con tolvaptán, así como un tercer paciente de un ensayo de ampliación abierto, mostraron aumentos en las enzimas hepáticas ( > 3 × LSN) con elevaciones concomitantes en BT ( > 2 × LSN). Notificación de Sospecha de Reacciones Adversas: Es importante notificar la sospecha de reacciones adversas al medicamento tras su autorización. Ello permite una supervisión continua de la relación beneficio/riesgo del medicamento. Se invita a los profesionales sanitarios a notificar las sospechas de reacciones adversas a través del Sistema Nacional de Farmacovigilancia al siguiente link: https://www.argentina.gob.ar/anmat/farmacovigilancia/ notificanos/eventosadversos y/o al Departamento de Farmacovigilancia de GADOR S.A. vía email a farmacovigilancia@gador.com o telefónicamente al 0800-220-2273(CARE). Siguiendo pautas internacionales, el producto TOBLAPTIN® se encuentra adherido a un Plan de Gestión de Riesgo (PGR) aprobado por la ANMAT. PGR: Conjunto de actividades e intervenciones en Farmacovigilancia diseñadas para identificar, caracterizar, prevenir o minimizar riesgos relacionados a productos medicinales, y la evaluación de la efectividad de esas intervenciones
Advertencias.
Necesidad urgente de elevar el sodio sérico de inmediato Tolvaptán no ha sido estudiado en un contexto de necesidad urgente de elevar el sodio sérico de inmediato. En estos pacientes, se debe considerar un tratamiento alternativo. Corrección excesivamente rápida del sodio sérico: Los pacientes con una concentración basal de sodio sérico demasiado baja pueden correr un mayor riesgo de sufrir una corrección excesivamente rápida de dicho valor. Una corrección excesivamente rápida de la hiponatremia (aumento 12 mmol/l/24 horas) puede causar una desmielinización osmótica que dé lugar a disartria, mutismo, disfagia, aletargamiento, cambios de tipo afectivo, cuadriparesia espástica, convulsiones, coma o muerte. Por consiguiente, una vez iniciado el tratamiento debe realizarse un seguimiento estrecho de la concentración de sodio sérico y del estado volémico de los pacientes (ver más arriba). Para reducir al mínimo el riesgo de que se produzca una corrección excesivamente rápida de la hiponatremia, el aumento del sodio sérico debe ser menor de 10-12 mmol/l/24 horas y de 18 mmol/l/48 horas. Por consiguiente, se aplican límites más conservadores durante la primera fase del tratamiento. En caso de que la corrección del sodio supere los 6 mmol/l durante las 6 primeras horas tras la administración o los 8 mmol/l durante las primeras 6-12 horas respectivamente, debe considerarse la posibilidad de que la corrección del sodio sérico se haya producido demasiado rápido. Debe realizarse una supervisión más frecuente del sodio sérico de estos pacientes y se recomienda administrarles fluidos hipotónicos. En caso de que el sodio sérico aumente a 12 mmol/l en un plazo de 24 horas o 18 mmol/l en un plazo de 48 horas, deberá interrumpirse o abandonarse el tratamiento con tolvaptán y administrarse a continuación fluidos hipotónicos. En el caso de los pacientes con un mayor riesgo de sufrir síndromes de desmielinización como por ejemplo los que presentan hipoxia, alcoholismo o desnutrición, el ritmo adecuado de la corrección del sodio puede ser inferior al de los pacientes que no presenten esos factores de riesgo. Deberá realizarse un seguimiento muy cuidadoso a estos pacientes. Los pacientes que hayan recibido otro medicamento o tratamiento para la hiponatremia que aumente la concentración de sodio en suero antes de iniciarse el tratamiento con TOBLAPTIN® deben tratarse con especial cuidado. Estos pacientes pueden correr un mayor riesgo de manifestar una corrección rápida de la concentración de sodio en suero durante los primeros 1-2 días de tratamiento debido a los potenciales efectos aditivos. No se recomienda la administración concomitante de TOBLAPTIN® con otros tratamientos y medicamentos para la hiponatremia que aumenten la concentración sérica de sodio durante el tratamiento inicial o en otros pacientes con una concentración basal de sodio sérico muy baja. Toxicidad hepática idiosincrática: Tolvaptán se ha asociado a elevaciones idiosincráticas de las concentraciones en sangre de alanina transaminasa (ALT) y de aspartato transaminasa (AST), con casos poco frecuentes de elevaciones concomitantes de la bilirrubina total (BT). En la experiencia posterior a la comercialización con tolvaptán en la PQRAD, se ha informado de insuficiencia hepática aguda que requiere trasplante de hígado. En un estudio doble ciego y controlado con placebo, en el que participaron pacientes con PQRAD, el periodo de manifestación de la lesión hepatocelular (por elevaciones de la ALT > 3 del LSN [límite superior normal]) se situó entre los 3 y 14 meses tras el comienzo del tratamiento y dichos aumentos fueron reversibles, dado que los valores de la ALT volvieron a ser < 3 del LSN, en un plazo de 1 a 4 meses. Si bien estas elevaciones concomitantes fueron reversibles al retirar inmediatamente la administración de tolvaptán, representan un potencial de daño hepático importante. Cambios similares con otros medicamentos se han asociado con posibles daños hepáticos irreversibles y potencialmente mortales. Los médicos prescriptores deben cumplir todas las medidas de seguridad que se indican a continuación. Con el fin de reducir el riesgo de que se produzca un daño hepático significativo y/o irreversible, se deben realizar análisis de sangre de transaminasas hepáticas y bilirrubina antes de iniciar el tratamiento con TOBLAPTIN®, así como cada mes durante 18 meses y a intervalos regulares (cada 3 meses) de ahí en adelante. Se recomienda monitorizar a la vez los síntomas que pueden ser indicativos de daño hepático, como fatiga, anorexia, náuseas, molestias en la parte superior derecha del abdomen, vómitos, fiebre, erupción cutánea, prurito, orina oscura o ictericia. En el caso de pacientes que presenten valores anómalos de ALT, AST o BT antes del inicio del tratamiento, y que cumplan los criterios para la interrupción permanente (ver más adelante), el uso de tolvaptán está contraindicado. En caso de presentar valores iniciales anómalos, por debajo de los límites para la interrupción permanente, solo se podrá iniciar el tratamiento si los beneficios potenciales del mismo superan a los posibles riesgos y se deben seguir realizando pruebas funcionales hepáticas con una mayor frecuencia. Se recomienda contar con el asesoramiento de un hepatólogo. Durante los primeros 18 meses de tratamiento, solo se podrá tratar con TOBLAPTIN® a aquellos pacientes cuyo médico haya determinado que la función hepática permite continuar con el tratamiento. En caso de manifestarse durante el tratamiento el comienzo de signos o síntomas sugestivos de una lesión hepática o aumentos anómalos clínicamente significativos de los valores de ALT o AST, se debe interrumpir inmediatamente la administración de TOBLAPTIN ® y repetir los análisis, lo que incluye la obtención, tan pronto como sea posible (idealmente en un plazo de 48 a 72 horas) de los valores de ALT, AST, BT y fosfatasa alcalina (FA). Los análisis se deben seguir realizando con una mayor frecuencia hasta que se resuelvan o estabilicen los síntomas/signos/analíticas, momento en el cual se puede reiniciar el tratamiento con TOBLAPTIN®. La práctica clínica actual sugiere que se debe interrumpir el tratamiento con TOBLAPTIN® una vez se confirme el aumento o alteración sostenida de las concentraciones de transaminasas, e interrumpirse definitivamente en caso de producirse aumentos significativos y/o si persisten los síntomas clínicos de daño hepático. Las directrices recomendadas para la interrupción permanente son: ALT o AST > 8 veces el LSN; ALT o AST > 5 veces el LSN durante más de 2 semanas; ALT o AST > 3 veces el LSN y BT > 2 veces el LSN o el Cociente Normalizado Internacional (RIN) > 1,5; ALT o AST > 3 veces el LSN con síntomas persistentes de daño hepático indicados anteriormente Si los valores de ALT y AST se mantienen por debajo de tres veces el LSN, se puede continuar el tratamiento con TOBLAPTIN® con precaución, manteniendo una supervisión frecuente con las mismas dosis o bien a dosis más bajas, ya que en algunos pacientes, las concentraciones de transaminasas parecen estabilizarse durante el tratamiento continuo. Disponibilidad de agua: Tolvaptán puede causar reacciones adversas relacionadas con la pérdida de agua, como sed, poliuria, nicturia, polaquiuria, xerostomía y deshidratación. Por consiguiente, los pacientes deben tener acceso a agua (o a otros líquidos acuosos) y deben ser capaces de beber estos líquidos en cantidad suficiente. Con el fin de evitar sufrir una sed excesiva o deshidratación, se debe indicar a los pacientes que beban en abundancia agua u otros líquidos acuosos al primer signo de sed. Además, los pacientes deben beber de 1 a 2 vasos de líquido antes de acostarse, con independencia de la sensación de sed que experimenten, y deben volver a beber tras cada micción nocturna. Si se trata con tolvaptán a pacientes con restricción de líquidos, se debe tener precaución especial para asegurarse de que los pacientes no se deshidraten en exceso. Deshidratación e hipovolemia: El tratamiento con tolvaptán causa acuaresis profusa, que normalmente se compensa parcialmente con la ingesta de líquidos. La deshidratación y la hipovolemia pueden ocurrir, especialmente en pacientes con depleción de volumen que reciben diuréticos o con restricción de líquidos. Se debe monitorizar la volemia en los pacientes tratados con tolvaptán, dado que este tratamiento puede dar lugar a una deshidratación grave, lo cual constituye un factor de riesgo de disfunción renal. En pacientes en tratamiento con tolvaptán que presentan signos o síntomas médicamente significativos de hipovolemia, se debe discontinuar o interrumpir el tratamiento con tolvaptán y proporcionar soporte médico con control de los signos vitales, balance hídrico y electrolitos. Se recomienda un control preciso del peso corporal. Una reducción progresiva del peso corporal podría ser un signo temprano de deshidratación progresiva. En caso de manifestarse una deshidratación, deberán tomarse las medidas adecuadas, incluyendo la necesidad de interrumpir o reducir la dosis de tolvaptán así como el aumento de la ingesta de líquidos. Se debe tener especial cuidado en el caso de pacientes con afecciones que impidan una ingesta adecuada de líquidos o que corran un mayor riesgo de deshidratación, por ejemplo aquellos con vómitos o diarrea. Obstrucción del flujo urinario: Hay que garantizar la excreción de orina. Los pacientes con una obstrucción parcial del flujo urinario, como por ejemplo los pacientes con hipertrofia de próstata o afectación de la micción, tienen un mayor riesgo de desarrollar retención aguda de orina. Equilibrio hídrico y electrolítico: Se debe supervisar en todos los pacientes el equilibrio hídrico y electrolítico, y especialmente en aquellos que presenten un trastorno renal o hepático. La administración de tolvaptán induce acuaresis copiosas y puede causar deshidratación, además de aumentar la concentración sérica de sodio, por lo que está contraindicada en pacientes con hipernatremia. Se tienen que evaluar los valores de creatinina sérica y de electrolitos así como los síntomas de desequilibrio electrolítico (por ejemplo, mareo, desmayo, palpitaciones, confusión, debilidad, inestabilidad de la marcha, hiperreflexia, convulsiones o coma) antes y después de iniciar el tratamiento con TOBLAPTIN®, para poder detectar una posible deshidratación. La administración de tolvaptán puede causar una elevación excesivamente rápida del sodio sérico ( > 12 mmol/l durante 24 horas); por consiguiente, se debe iniciar la supervisión de las concentraciones de sodio sérico en todos los pacientes como máximo 4-6 horas después de iniciarse el tratamiento