OFANIR®
ADIUM
Agente inhibidor de la proteína tirosina quinasa.
Composición.
Cada cápsula con contenido líquido de 100 mg contiene: Nintedanib etanosulfonato 120,400 mg Equivalente a Nintedanib 100,000 mg. Cada cápsula con contenido líquido de 150 mg contiene: Nintedanib etanosulfonato 180,600 mg Equivalente a Nintedanib 150,000 mg.
Farmacología.
Mecanismo de acción: Nintedanib es un inhibidor de la tirosina quinasa cuya acción comprende a los receptores del factor de crecimiento derivado de plaquetas (PDGFR) y, los receptores del factor de crecimiento de fibroblastos (FGFR) 1-3 y los receptores de factor de crecimiento endotelial vascular (VEGFR) 1-3. Nintedanib se une competitivamente al sitio de unión a ATP de estos receptores y bloquea la señalización intracelular que es crucial para la proliferación, migración y transformación de los fibroblastos, que constituyen los mecanismos esenciales de la patología de la FPI. Además, nintedanib inhibe las quinasas Flt-3 (proteína tirosina quinasa similar a Fms), Lck (proteína tirosina quinasa específica de linfocitos), Lyn (proteína tirosina quinasa Lyn) y Src (proteína tirosina quinasa proto-oncogénica Src). Efectos farmacodinámicos: Nintedanib inhibe la activación de las cascadas de señalización FGFR y PDGFR, que están estrechamente implicadas en la proliferación, la migración y la diferenciación de los fibroblastos/miofibroblastos pulmonares, las células características en la patología de la fibrosis pulmonar idiopática. En la actualidad, no se ha logrado aclarar por completo el impacto potencial de la inhibición del VEGFR provocada por nintedanib ni la actividad anti-angiogénica de nintedanib en la patología de la FPI. En modelos preclínicos de fibrosis pulmonar, nintedanib ejerce una potente acción antifibrótica y antinflamatoria. Nintedanib inhibe la proliferación, la migración y la transformación de fibroblastos en miofibroblastos de los fibroblastos pulmonares humanos de pacientes con FPI. Farmacocinética: Absorción: Nintedanib alcanza la concentración plasmática máxima aproximadamente de 2 a 4 horas después de la administración oral junto con alimentos (rango de 0,5 a 8 horas). La biodisponibilidad absoluta de una dosis de 100 mg es del 4,69% en voluntarios sanos. La absorción y la biodisponibilidad disminuyen por los efectos de los transportadores y por el metabolismo sustancial de primer paso hepático. La proporcionalidad de la dosis se demostró mediante un aumento de la exposición a nintedanib (rango de dosis de 50 a 450 mg una vez al día y de 150 a 300 mg dos veces al día). Las concentraciones plasmáticas en estado estacionario se logran, a más tardar, en el plazo de una semana de dosificación. Después de la ingesta de alimentos, la exposición a nintedanib aumenta en aproximadamente el 20% en comparación con la administración en ayunas y la absorción se retrasa (mediana de tmax en ayunas: 2,00 h; con alimentos: 3,98 h). Distribución: Nintedanib sigue al menos una cinética de distribución bifásica. Después de una infusión intravenosa, se observa un alto volumen de distribución (Vss: 1.050 litro). La unión a proteínas in vitro en el plasma humano es alta, con una fracción unida del 97,8%. Se considera que la albúmina sérica es la proteína de unión más importante. Nintedanib se distribuye preferentemente en el plasma, con una relación sangre/plasma de 0,869. Biotransformación: La reacción metabólica prevalente para nintedanib es la ruptura hidrolítica mediante esterasas que dan lugar a la fracción de ácido libre BIBF 1202. A continuación, BIBF 1202 se glucuronida mediante enzimas uridina 5'-difosfo-glucurosiltransferasa (UGT), concretamente UGT 1A1, UGT 1A7, UGT 1A8 y UGT 1A10, a glucurónido de BIBF 1202. Tan solo una pequeña proporción de la biotransformación de nintedanib se realiza a través de las vías CYP, siendo CYP 3A4 la enzima predominante implicada. El principal metabolito dependiente de CYP no pudo detectarse en el plasma en un estudio farmacocinético realizado con humanos. In vitro, el metabolismo dependiente de CYP representa aproximadamente un 5% en comparación con aproximadamente un 25% de ruptura de ésteres. Nintedanib, BIBF 1202 y el glucurónido BIBF 1202 no inhibieron ni indujeron las enzimas CYP en animales. Por lo tanto, no cabe esperar interacciones farmacológicas entre nintedanib y sustratos de CYP, inhibidores de CYP o inductores de CYP. Eliminación: El aclaramiento plasmático total después de la infusión intravenosa es alto (aclaramiento: 1.390 ml/min). La eliminación por la orina del principio activo inalterado en el plazo de 48 horas es de aproximadamente el 0,05% de la dosis después de la administración oral, y de aproximadamente 1,4% de la dosis después de la administración intravenosa; el aclaramiento renal es de 20 ml/min. La principal vía de eliminación del fármaco marcado radioactivamente después de la administración oral de [C14]-nintedanib es la excreción biliar/fecal (93,4% de la dosis). La contribución de la eliminación renal al aclaramiento total es baja (0,649% de la dosis). La recuperación total se considera completa (por encima del 90%) en los cuatro días posteriores a la dosificación. La vida media terminal de nintedanib es de entre 10 y 15 horas. Linealidad/No linealidad: La farmacocinética de nintedanib se puede considerar lineal respecto al tiempo (es decir, los datos de una sola dosis pueden extrapolarse a los datos de múltiples dosis). La acumulación en el caso de múltiples administraciones es de 1,04 veces para la Cmax y de 1,38 veces para el AUC. Las concentraciones mínimas de nintedanib permanecen estables durante más de un año. Transporte: Nintedanib es un sustrato de la gp-P. Se demostró que nintedanib no es un sustrato ni un inhibidor de OATP-1B1, OATP-1B3, OATP-2B1, OCT-2 o MRP-2 in vitro. Nintedanib tampoco resultó ser un sustrato de la BCRP. Solo se observó in vitro un leve potencial inhibidor en el OCT-1, la BCRP y la gp-P, pero se considera que esto tiene una baja relevancia clínica. Lo mismo se aplica a nintedanib como sustrato del OCT-1. Análisis farmacocinético poblacional en poblaciones especiales: Las propiedades farmacocinéticas de nintedanib fueron similares entre voluntarios sanos, pacientes con FPI y pacientes con cáncer. Basándose en los resultados de un análisis farmacocinético poblacional en pacientes con FPI y cáncer de pulmón no microcítico (CPNM) y en las investigaciones descriptivas, la exposición a nintedanib no se ve afectada por el sexo (corregido por el peso corporal), la insuficiencia renal leve y moderada (calculada según el aclaramiento de creatinina), el consumo de alcohol ni por el genotipo de la gp-P. Los análisis farmacocinéticos poblacionales indicaron efectos moderados en la exposición a nintedanib dependiendo de la edad, el peso corporal y la raza. Teniendo en cuenta la alta variabilidad de la exposición entre individuos, los efectos moderados observados no se consideran clínicamente relevantes. Edad: La exposición a nintedanib aumenta linealmente con la edad. El AUC disminuye en un 16% en el caso de un paciente de 45 años y aumenta en un 13% en el caso de un paciente de 76 años en comparación con un paciente con la mediana de edad de 62 años. Basándose en un modelo farmacocinético poblacional, se observó un aumento en la exposición a nintedanib de aproximadamente entre el 20 y el 25% en pacientes de 75 años o más, en comparación con pacientes menores de 65 años. No se han realizado estudios con poblaciones pediátricas. Peso corporal: Se observa una correlación inversa entre el peso corporal y la exposición a nintedanib. El AUC aumenta en un 25% en el caso de un paciente de 50 kg (percentilo 5) y disminuye en un 19% en el caso de un paciente de 100 kg (percentilo 95) en comparación con un paciente con la mediana de peso de 71,5 kg. Raza: La exposición media de la población a nintedanib es un 33-50% superior en pacientes chinos, taiwaneses e indios y un 16% superior en pacientes japoneses, mientras que es un 16-22% inferior en el caso de pacientes coreanos comparados con los caucásicos (corregido por peso corporal). Los datos procedentes de individuos de raza negra fueron muy limitados, pero se encuentran en el mismo rango que en el caso de los caucásicos. Insuficiencia hepática:La exposición a nintedanib basándose en la Cmax y el AUC es 2,2 veces más alta en voluntarios con insuficiencia hepática leve (Child Pugh A) y 7,6 veces más alta en voluntarios con insuficiencia hepática moderada (Child Pugh B), comparado con los voluntarios sanos. No se han realizado estudios en individuos con insuficiencia hepática grave (Child Pugh C). Tratamiento conjunto con pirfenidona: De acuerdo con los resultados de un estudio farmacocinético específico, no existe evidencia de una interacción farmacocinética relevante entre nintedanib y pirfenidona cuando se administran en combinación.
Indicaciones.
OFANIR está indicado para el tratamiento de la fibrosis pulmonar idiopática (FPI) en pacientes adultos.
Dosificación.
Posología: El tratamiento con OFANIR debe ser iniciado y supervisado por médicos con experiencia en el diagnóstico y el tratamiento de la FPI. La dosis recomendada de OFANIR es de 150 mg dos veces al día con un intervalo de aproximadamente 12 horas entre sí. Forma de administración: Las cápsulas de OFANIR deben tomarse por vía oral, preferentemente con alimentos. Deben tragarse enteras con agua y no deben masticarse ni triturarse. No debe excederse la dosis diaria máxima recomendada de 300 mg. Ajustes de la dosis: Además de la instauración de tratamiento sintomático, en el caso de corresponder, el manejo de los efectos secundarios de OFANIR (véase Advertencias y Reacciones adversas) podría incluir la reducción de la dosis y la interrupción temporaria de la administración del fármaco hasta que la reacción adversa en cuestión se haya resuelto a un nivel que permita la continuación del tratamiento. El tratamiento con OFANIR podrá reanudarse en la dosis completa (150 mg dos veces al día) o en una dosis reducida (100 mg dos veces al día). Si el paciente no tolera el régimen de 100 mg dos veces al día, debe discontinuarse el tratamiento con OFANIR. En el caso de interrupciones a raíz de elevaciones de la transaminasa (AST o ALT) > 3 veces el límite superior del rango normal (ULN), una vez que las transaminasas hayan retornado a los valores basales, el tratamiento con OFANIR podrá reiniciarse en una dosis reducida (100 mg dos veces al día), que luego podrá incrementarse hasta llegar a la dosis completa (150 mg dos veces al día). Población pediátrica: No se recomienda su uso en pacientes pediátricos. Pacientes de edad avanzada (65 años): No se requieren ajustes de dosis sobre la base de la edad del paciente. Los pacientes con una edad igual o superior a 75 años pueden tener más probabilidades de necesitar una reducción de la dosis para tratar los efectos adversos. Raza y peso corporal: Basándose en los análisis farmacocinéticos (FC) poblacionales, a priori no es necesario realizar ajustes de la dosis de nintedanib. Los datos de seguridad para pacientes de raza negra y afroamericana son limitados. Insuficiencia renal: Menos del 1 % de una dosis única de nintedanib se elimina a través del riñón. No se requiere un ajuste de la dosis inicial en los pacientes con insuficiencia renal leve a moderada. La seguridad, la eficacia y la farmacocinética de nintedanib no han sido estudiadas en pacientes con insuficiencia renal grave (aclaramiento de creatinina < 30 ml/min). Insuficiencia hepática: Nintedanib se elimina principalmente a través de la excreción biliar y fecal ( > 90 %). Hay un aumento de la exposición en pacientes con insuficiencia hepática (Child Pugh A, Child Pugh B). En pacientes con insuficiencia hepática leve (Child Pugh A), la dosis recomendada de OFANIR es 100 mg dos veces al día con un intervalo de aproximadamente 12 horas entre sí. Debe considerarse la interrupción o suspensión del tratamiento para el manejo de las reacciones adversas, en pacientes con insuficiencia hepática leve (Child Pugh A). La seguridad y la eficacia de nintedanib no han sido investigadas en pacientes con insuficiencia hepática clasificada como Child Pugh B o C. No se recomienda el tratamiento con OFANIR en los pacientes con insuficiencia hepática moderada (Child Pugh B) o grave (Child Pugh C).
Contraindicaciones.
Pacientes con hipersensibilidad conocida a nintedanib, o a la soja, o a cualquiera de sus excipientes. Embarazo.
Reacciones adversas.
Los datos de seguridad que se brindan a continuación están basados estudios publicados de pacientes con Fibrosis Pulmonar Idiopática (FPI) tratados con nintedanib 150 mg dos veces al día frente a placebo durante 52 semanas y también se basan en los datos observados durante el período posterior a la comercialización. Los eventos adversos asociados con el uso de nintedanib informados con mayor frecuencia fueron: diarrea, náuseas y vómitos, dolor abdominal, disminución del apetito, descenso de peso y elevación de las enzimas hepáticas. Para el manejo de las reacciones adversas seleccionadas, ver también Advertencias y Precauciones. La tabla siguiente incluye un resumen de las reacciones adversas según la clasificación de órganos del sistema MedDRA y la categoría de frecuencia. Las categorías de frecuencia se definen utilizando la siguiente convención: Muy frecuentes (1/10), frecuentes (1/100 a < 1/10), poco frecuentes (1/1.000 a < 1/100), raras (1/10.000 a < 1/1.000), muy raras ( < 1/10.000), frecuencia no conocida (no se puede estimar a partir de los datos disponibles). Dentro de cada grupo de frecuencia, las reacciones adversas se presentan por orden descendente de gravedad.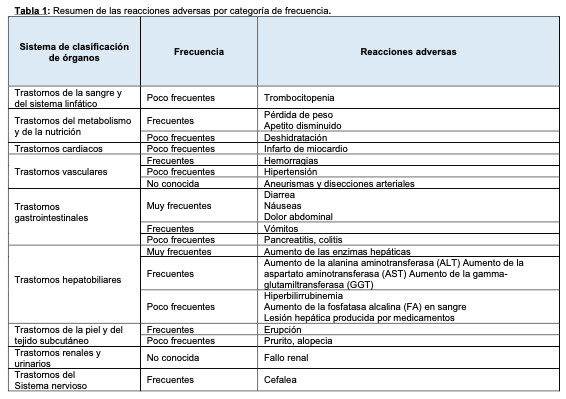
Notificación de sospechas de reacciones adversas: Es importante notificar sospechas de reacciones adversas al medicamento tras su autorización. Ello permite una supervisión continuada de la relación beneficio/riesgo del medicamento. Se invita a los profesionales sanitarios a notificar las sospechas de reacciones adversas a través del Departamento de Farmacovigilancia del laboratorio vía email: fvigilancia@raffo.com.ar o a través de los teléfonos (011) 4509-7100/7127, y/o del Sistema Nacional de Farmacovigilancia al siguiente link: http://sistemas.anmat.gov.ar/aplicaciones_net/applications/fvg_eventos_adversos_nuevo/index.html
Precauciones.
Población pediátrica: La seguridad y la eficacia de Nintedanib en pacientes pediátricos no han sido estudiadas en estudios clínicos. No se recomienda su uso en pacientes pediátricos. Pacientes de edad avanzada (≥ 65 años): No se observaron diferencias en general en lo que respecta a la seguridad y la eficacia en los pacientes de edad avanzada en comparación con los pacientes menores de 65 años de edad. Insuficiencia renal: Menos del 1% de una dosis única de nintedanib se excreta a través de los riñones. La seguridad, la eficacia y la farmacocinética de nintedanib no han sido estudiadas en pacientes con insuficiencia renal grave (clerance de creatinina:CrCl < 30 ml/min). Insuficiencia hepática: Nintedanib se elimina primordialmente a través de la excreción por vía biliar/fecal ( > 90%.). La exposición aumenta en pacientes con insuficiencia hepática Child Pugh A, Child Pugh B. La seguridad y la eficacia de nintedanib no han sido estudiadas en pacientes con insuficiencia hepática moderada (Child Pugh B) o grave (Child Pugh C). Por lo tanto, no se recomienda el tratamiento con nintedanib en dichos pacientes. Sobre la base de que existe una mayor exposición, es posible que los pacientes con insuficiencia hepática leve (Child Pugh A) corran más riesgos de sufrir eventos adversos. Los pacientes con insuficiencia hepática leve (Child Pugh A) deben tratarse con una dosis reducida de nintedanib (ver Posología y advertencias). Poblaciones especiales: En un estudio en pacientes con cáncer de pulmón no microcítico, la frecuencia de efectos adversos graves en pacientes tratados con nintedanib más docetaxel fue mayor en los pacientes con un peso corporal inferior a 50 kg que en los pacientes con un peso ≥ 50 kg. No obstante, el número de pacientes con un peso corporal inferior a 50 kg fue reducido. Así pues, se recomienda realizar un control estrecho en el caso de pacientes que pesen < 50 kg. Interacciones: Los estudios de interacción se han realizado únicamente en adultos. Glucoproteína P (P-gp): Nintedanib es un sustrato de la P-gp. La coadministración con ketoconazol, un inhibidor potente de la P-gp, incrementa la exposición a nintedanib por un factor de 1,61 sobre la base del AUC y por un factor de 1,83 sobre la base de la Cmáx. La coadministración con rifampicina, un potente inductor de la P-gp, reduce la exposición a nintedanib a un 50,3% sobre la base del AUC y a un 60,3% sobre la base de la Cmáx. Si se coadministran inhibidores potentes de la P-gp (p. ej., ketoconazol o eritromicina o ciclosporina) junto con nintedanib, puede incrementarse la exposición a nintedanib. En tales casos, debe implementarse un control estrecho de los pacientes a fin de determinar la tolerancia a nintedanib. El manejo de los efectos secundarios puede requerir la reducción de la dosis o bien la suspensión temporaria o definitiva del tratamiento con nintedanib. Los inductores potentes de la P-gp (p. ej., rifampicina, carbamazepina, fenitoína y hierba de San Juan) pueden reducir la exposición a nintedanib. La administración conjunta con nintedanib se debe valorar cuidadosamente. Alimentos: Se recomienda que nintedanib se administre con alimentos. Enzimas del citocromo (CYP): Sólo una pequeña parte de la biotransformación de nintedanib involucra las vías del CYP. Nintedanib y sus metabolitos, la fracción ácido libre BIBF 1202 y su glucurónido BIBF 1202 glucurónido, no inhiben ni inducen a las enzimas del CYP, por lo tanto, se considera que la probabilidad de que se produzcan interacciones medicamentosas con nintedanib basadas en el metabolismo del CYP es baja. Coadministración con otros fármacos: La administración conjunta de nintedanib con docetaxel (75 mg/m2) no afectó significativamente a la farmacocinética de estos medicamentos. No se evaluó el potencial de interacciones de nintedanib con los anticonceptivos hormonales. Fertilidad: Sobre la base de las investigaciones preclínicas, no existe evidencia de que este fármaco afecte la fertilidad masculina. No se dispone de datos con humanos o animales sobre los potenciales efectos de nintedanib en la fertilidad femenina. Mujeres en edad fértil/Anticoncepción: Nintedanib puede causar daño fetal en humanos. Se debe advertir a las mujeres en edad fértil y con potencial para procrear que sean tratadas con nintedanib que eviten quedar embarazadas mientras estén tomando este medicamento, utilizando métodos anticonceptivos adecuados durante el tratamiento y hasta cumplidos al menos 3 meses desde la última dosis de nintedanib. Dado que no se ha estudiado el efecto de nintedanib sobre el metabolismo y la eficacia de los anticonceptivos hormonales, para evitar un embarazo deberá utilizarse doble método anticonceptivo; es decir un método de barrera adicional Embarazo: No existe información sobre el uso de nintedanib en las mujeres embarazadas; sin embargo, los estudios preclínicos en animales han confirmado la toxicidad para la reproducción. Dado que nintedanib puede causar daño fetal también en los seres humanos, este fármaco no debe ser utilizado durante el embarazo. Es necesario realizar una prueba de embarazo como mínimo antes del inicio del tratamiento con nintedanib. Se debe indicar a las pacientes que deben notificar a su médico de inmediato si quedaran embarazadas durante el tratamiento con nintedanib. Si la paciente quedara embarazada mientras esté recibiendo nintedanib, deberá ser asesorada sobre el potencial riesgo para el feto. Debe considerarse la interrupción del tratamiento. Lactancia: No existe información sobre la excreción de nintedanib y sus metabolitos en la leche humana. Los estudios preclínicos han indicado que se segregaron pequeñas cantidades de nintedanib y sus metabolitos (≤ 0,5% de la dosis administrada) en la leche de las ratas en período de lactancia. No se puede excluir la posibilidad de un riesgo para los neonatos/lactantes. Debe suspenderse la lactancia durante el tratamiento con nintedanib. Efectos sobre la capacidad para conducir vehículos y operar maquinaria: La influencia de nintedanib sobre la capacidad para conducir y utilizar máquinas es pequeña. Debe indicarse a los pacientes que deben tener precaución al conducir vehículos u operar maquinaria durante el tratamiento con nintedanib. Excipientes: Las cápsulas de nintedanib contienen lecitina de soja. Este medicamento es libre de gluten y lactosa.
Advertencias.
Trastornos gastrointestinales: La diarrea fue el evento gastrointestinal más frecuente en los estudios con nontedanib. En la mayoría de los pacientes, el evento fue de intensidad leve a moderada y se produjo dentro de los primeros 3 meses de tratamiento. La diarrea condujo a una reducción de la dosis y a la discontinuación del nintedanib en algunos pacientes. La diarrea debe tratarse ante la aparición de los primeros síntomas con una hidratación adecuada y con productos medicinales antidiarreicos, como loperamida, y puede requerir la interrupción del tratamiento. El tratamiento con nintedanib podrá reanudarse en una dosis reducida (100 mg dos veces al día) o en la dosis completa (150 mg dos veces al día). En el caso de que persista un cuadro de diarrea grave a pesar del tratamiento sintomático, deberá discontinuarse el tratamiento con nintedanib Las náuseas y los vómitos fueron eventos adversos informados con frecuencia en los estudios clínicos. En la mayoría de los pacientes con náuseas y vómitos, el evento fue de intensidad leve a moderada. Las náuseas y los vómitos condujeron a la discontinuación de nintedanib en algunos pacientes. Si los síntomas persisten a pesar de haberse instaurado un tratamiento de soporte adecuado (lo que incluye terapia antiemética), puede ser necesario implementar una reducción de la dosis o la interrupción del tratamiento. El tratamiento podrá reanudarse en una dosis reducida (100 mg dos veces al día) o en la dosis completa (150 mg dos veces al día). Ante la presencia de síntomas graves que persistan, deberá discontinuarse el tratamiento con nintedanib. La diarrea y los vómitos pueden producir deshidratación y/o desequilibrio electrolítico. Neutropenia y sepsis: En los pacientes con cáncer de pulmón no microcítico, tratados con nintedanib en combinación con docetaxel la frecuencia de neutropenia de grado 3 según los CTCAE fue mayor que en los pacientes tratados con docetaxel en monoterapia. Se han observado complicaciones asociadas, como la sepsis o la neutropenia febril. En el caso de pacientes que reciben el tratamiento de nintedanib en combinación con docetaxel es necesario realizar un control frecuente de los recuentos celulares sanguíneos completos al principio de cada ciclo de tratamiento y alrededor de la fecha nadir, así como siempre que esté clínicamente indicado después de administrar el último ciclo de la combinación. Función hepática: Se han observado casos de lesión hepática producida por medicamentos con el tratamiento con nintedanib. En el período posterior a la comercialización, se han informado casos graves y casos no graves de daño hepático causado por el medicamento, incluso daño hepático grave con desenlace mortal. La mayoría de los eventos hepáticos ocurren dentro de los primeros tres meses de tratamiento. Por lo tanto, deben determinarse los niveles de bilirrubina y transaminasas hepáticas al iniciarse el tratamiento con nintedanib, a intervalos periódicos durante los primeros tres meses de tratamiento y luego a intervalos periódicos (p. ej., en cada visita del paciente) o según esté clínicamente indicado. Las elevaciones de las enzimas hepáticas (ALT, AST, ALKP, gamma glutamiltransferasa (GGT)) y de los valores de bilirrubina fueron reversibles con la reducción de la dosis o la interrupción del tratamiento, en la mayoría de los casos. En el caso de detectarse elevaciones de las transaminasas (AST o ALT) > 3 veces el límite superior del rango normal (ULN), se recomienda la reducción de la dosis o la interrupción del tratamiento con nintedanib y el monitoreo estrecho del paciente. Una vez que las transaminasas hayan retornado a los valores basales, podrá incrementarse nuevamente la dosis (150 mg dos veces al día) o bien reiniciarse el tratamiento con nintedanib en una dosis reducida (100 mg dos veces al día), que luego podrá incrementarse hasta llegar a la dosis completa (150 mg dos veces al día). Si alguna de estas elevaciones en los parámetros de función hepática estuviera asociada con signos o síntomas clínicos de lesión hepática, como ictericia, deberá interrumpirse en forma definitiva el tratamiento con nintedanib. Deben investigarse las posibles causas alternativas de las elevaciones de las enzimas hepáticas. Los pacientes con bajo peso corporal ( < 65 kg), los de raza asiática y las mujeres tienen un mayor riesgo de elevaciones de las enzimas hepáticas. La exposición a nintedanib se incrementa de manera lineal en función de la edad de los pacientes, lo que también puede dar lugar a un mayor riesgo de desarrollar elevaciones de las enzimas hepáticas. Se recomienda un monitoreo estrecho en los pacientes que presenten estos factores de riesgo. Función renal: Se han notificado casos de insuficiencia/fallo renal, algunos de ellos con un desenlace mortal, con el uso de nintedanib. Los pacientes deben ser controlados durante el tratamiento con nintedanib, con especial atención aquellos pacientes que presenten factores de riesgo para insuficiencia/fallo renal. En caso de insuficiencia/fallo renal, se debe considerar el ajuste del tratamiento. Hemorragia: La inhibición del VEGFR podría estar asociada con un mayor riesgo de sangrado. En los estudios con nintedanib, la epistaxis no grave fue el evento de sangrado más frecuente. Los pacientes con riesgo conocido de sangrado, no fueron incluidos en dichos estudios, por lo tanto, el tratamiento con nintedanib en estos pacientes podrá implementarse únicamente en el caso de que el beneficio previsto supere el potencial riesgo implicado. En el período posterior a la comercialización, se han observado eventos hemorrágicos graves y no graves, algunos de los cuales resultaron mortales. En caso de sangrado, se debe considerar el ajuste de la dosis, la interrupción o la suspensión del tratamiento basándose en la evaluación clínica. Los episodios de sangrado pueden afectar, entre otros, a los sistemas gastrointestinal, respiratorio y nervioso central, si bien los más frecuentes son gastrointestinales. Anticoagulación terapéutica: No existen datos de ensayos clínicos relativos a pacientes con una predisposición hereditaria al sangrado ni relativos a pacientes que reciben una dosis completa de anticoagulante antes de iniciar el tratamiento con nintedanib. En el caso de pacientes que seguían un tratamiento crónico con bajas dosis de heparinas de bajo peso molecular o de ácido acetilsalicílico, no se observó un aumento de la frecuencia de hemorragias. Los pacientes que desarrollaron episodios tromboembólicos durante el tratamiento y que requirieron un tratamiento anticoagulante pudieron continuar el tratamiento con nintedanib y no mostraron un aumento en la frecuencia de episodios hemorrágicos. Los pacientes que tomen anticoagulantes de forma conjunta, tales como la warfarina o el fenprocumon, se deben controlar de forma regular para ver si se producen cambios en el tiempo de protrombina, la Razón Internacional Normalizada (RIN) y/o episodios hemorrágicos clínicos. Metástasis cerebral: - Metástasis cerebral estable: No se observó un aumento de la frecuencia de hemorragia cerebral en pacientes con metástasis cerebrales pretratadas de forma adecuada que se habían mantenido estables durante ≥ 4 semanas antes de comenzar el tratamiento con nintedanib en estudios cáncer de pulmón no microcítico. No obstante, es preciso controlar de forma estrecha a tales pacientes para ver si se producen signos y síntomas de hemorragia cerebral. - Metástasis cerebral activa: Los pacientes con metástasis cerebral activa se excluyeron de los ensayos clínicos, por lo que no se recomienda tratarlos con nintedanib. Eventos tromboembólicos arteriales: Los pacientes con antecedentes recientes de infarto de miocardio o accidente cerebrovascular fueron excluidos de los estudios con nintedanib. No obstante, se observó un aumento de la frecuencia de episodios tromboembólicos arteriales en pacientes con fibrosis pulmonar idiopática (FPI) tratados con nintedanib en monoterapia. Se deben tomar las debidas precauciones cuando se trate a pacientes con un alto riesgo cardiovascular, incluyendo una enfermedad conocida de las arterias coronarias. En pacientes que desarrollan signos o síntomas de isquemia miocárdica aguda, se debe valorar la necesidad de interrumpir el tratamiento. Aneurismas y disecciones arteriales: El uso de inhibidores de la vía VEGF en pacientes con o sin hipertensión puede promover la formación de aneurismas y/o disecciones arteriales. Antes de iniciar el tratamiento con nintedanib, este riesgo se debe evaluar de forma cuidadosa en pacientes con factores de riesgo como hipertensión o antecedentes de aneurisma. Tromboembolismo venoso: Los pacientes con cáncer de pulmón no microcítico que fueron tratados con nintedanib presentaron un mayor riesgo de sufrir un tromboembolismo venoso, incluyendo la embolia pulmonar y la trombosis venosa profunda, no así en los pacientes tratados por fibrosis pulmonar. Es preciso controlar de forma estrecha a tales pacientes para ver si se producen episodios tromboembólicos. Se debe tener precaución especialmente en pacientes con factores de riesgo adicionales de episodios tromboembólicos. El tratamiento con nintedanib se debe suspender en el caso de pacientes con reacciones tromboembólicas venosas que puedan poner en peligro su vida. Perforaciones gastrointestinales: Debido a su mecanismo de acción, los pacientes tratados con nintedanib podrían tener un mayor riesgo de padecer eventos de perforación gastrointestinal. Se han informado casos de perforaciones gastrointestinales, algunos de ellos mortales, en el período posterior a la comercialización. Debe tenerse especial cuidado al tratar a pacientes con una cirugía abdominal previa o antecedentes recientes de perforación de un órgano hueco, antecedentes de úlcera péptica, enfermedad diverticular o que estén recibiendo tratamiento concomitante con corticosteroides o AINE. Por lo tanto, debe dejarse transcurrir un mínimo de 4 semanas luego de una cirugía mayor, lo que incluye una cirugía abdominal, antes de iniciar la administración de nintedanib. El tratamiento con nintedanib debe suspenderse definitivamente en los pacientes que desarrollen una perforación gastrointestinal. Hipertensión: La administración de nintedanib puede aumentar la presión arterial. La presión arterial sistémica se debe medir de forma periódica y siempre que esté indicado clínicamente. Complicaciones en la cicatrización de las heridas: No se observó un aumento de la frecuencia de problemas de cicatrización de las heridas en los estudios clínicos. Sobre la base de su mecanismo de acción, nintedanib podría dificultar la normal cicatrización de las heridas. No se llevó a cabo ningún estudio específico en el que se investigara el efecto de nintedanib sobre la cicatrización de las heridas. Por lo tanto, el tratamiento con nintedanib debe ser iniciado, o reanudado en el caso de haber sido suspendido por una intervención quirúrgica, tras la confirmación de una correcta cicatrización de las heridas sobre la base del criterio clínico. Administración conjunta con pirfenidona: El tratamiento conjunto de nintedanib con pirfenidona se evaluó en un estudio farmacocinético. De acuerdo con estos resultados, no existe evidencia de una interacción farmacocinética relevante entre nintedanib y pirfenidona cuando se administran en combinación. Teniendo en cuenta el número limitado de pacientes, este estudio detectó solo las reacciones adversas más frecuentes y mostró un aumento de las reacciones adversas gastrointestinales y una tendencia a un aumento de las reacciones adversas hepáticas. Dada la similitud de los perfiles de seguridad de ambos medicamentos, cabe prever reacciones adversas aditivas, incluidas reacciones adversas gastrointestinales y hepáticas. No se ha establecido el balance beneficio-riesgo del tratamiento conjunto con pirfenidona. Efecto en el intervalo QT: En los estudios realizados, no se observó ninguna evidencia de una prolongación del intervalo QT al administrar nintedanib. Como se sabe que algunos otros inhibidores de la tirosina quinasa ejercen un efecto sobre el intervalo QT, se debe tomar precaución cuando se administre nintedanib a pacientes que puedan desarrollar una prolongación del intervalo QTc. Reacción alérgica: Se sabe que los productos alimenticios con soja provocan reacciones alérgicas, incluido un shock anafiláctico grave en personas con alergia a la soja. Los pacientes con una alergia conocida a la proteína del maní presentan un mayor riesgo de desarrollar reacciones graves a los preparados de soja.
Conservación.
A temperatura ambiente entre 15° C y 30° C.
Sobredosificación.
No existe ningún antídoto ni tratamiento específico para la sobredosis de nintedanib. La dosis única más alta de nintedanib administrada en los estudios clínicos fue 450 mg una vez al día. Asimismo, 2 pacientes tuvieron una sobredosis de un máximo de 600 mg dos veces al día durante un total de hasta ocho días. Los eventos adversos observados fueron concordantes con el perfil de seguridad conocido de nintedanib, es decir, elevación de las enzimas hepáticas y síntomas gastrointestinales. Ambos pacientes se recuperaron de dichas reacciones adversas. En un paciente que fue expuesto inadvertidamente a una dosis de 600 mg diarios durante un total de 21 días, se produjo un evento adverso no grave (nasofaringitis), el cual se resolvió durante el período de administración de la dosis incorrecta, sin que se observara el inicio de otros eventos informados. En el caso de producirse una sobredosis, es preciso interrumpir el tratamiento e iniciar las medidas de apoyo generales que resulten adecuadas. Ante la eventualidad de una sobredosificación, concurrir al Hospital más cercano o comunicarse con los Centros de Toxicología: Hospital de Pediatría Ricardo Gutiérrez: Teléfono: (011) 4962-6666/2247, Hospital A. Posadas: Teléfono: (011) 4654-6648/4658-7777. Centro de Asistencia Toxicológica de La Plata: Teléfono: (0221) 451-5555.
Presentación.
Envases conteniendo 60 y 120 cápsulas con contenido líquido.
Revisión.
18/08/2020.