TOLVAR
BAGO
Inmunosupresor. Inmunosupresor selectivo. Inhibidor de las enzimas Janus quinasas.
Composición.
Cada Comprimido Recubierto contiene: Tofacitinib (como Tofacitinib Citrato) 5 mg. Excipientes: Lactosa 62 mg; Carboximetilcelulosa Reticulada 6 mg; Estearato de Magnesio 2 mg; Celulosa Microcristalina c.s.p. 200 mg; Opadry II YS-30-18056 White (*1) 9 mg; Opadry II YS-19-19054 Clear (*2) 1 mg. (*1) Opadry II YS-30-18056 White está compuesto por Lactosa; Hipromelosa; Dióxido de Titanio; Triacetina. (*2) Opadry II YS-19-19054 Clear está compuesto por Hipromelosa; Maltodextrina; Triacetina. Este Medicamento es Libre de Gluten.
Farmacología.
Acción farmacológica: Mecanismo de acción: Tofacitinib es un inhibidor de las Janus quinasas (JAK, por sus siglas en inglés). Las JAK son enzimas intracelulares que transmiten señales derivadas de las interacciones de las citocinas o del receptor del factor de crecimiento sobre la membrana celular para influir en los procesos celulares de hematopoyesis y la función celular inmune. Dentro de la vía de señalización, las JAK fosforilan y activan los transductores de señal y activadores de la transcripción (STAT, por sus siglas en inglés) que modulan la actividad intracelular, incluyendo la expresión génica. Tofacitinib modula la vía de señalización en la etapa de las JAK, impidiendo la fosforilación y activación de los STAT. Las enzimas JAK transmiten la señal de las citocinas mediante la combinación por pares de JAK (por ejemplo, JAK1/JAK3, JAK1/JAK2, JAK1/TyK2, JAK2/JAK2). Tofacitinib inhibió la actividad in vitro de los pares JAKI/JAK2, JAK1/JAK3 y JAK2/JAK2 con IC50 de 406, 56 y 1377 nM, respectivamente. Sin embargo, se desconoce la relevancia de combinaciones específicas de JAK para la eficacia terapéutica. Farmacodinamia: El tratamiento con Tofacitinib se asoció con reducciones dosis-dependientes en el número de linfocitos "natural killers" CD16/56+ circulantes, con reducciones máximas estimadas que se producen después de aproximadamente 8 a 10 semanas de comenzado el tratamiento. Estos cambios generalmente se revierten después de 2 a 6 semanas de la suspensión del tratamiento. El tratamiento con Tofacitinib se asoció con aumentos dosis-dependientes en los recuentos de células B. Los cambios en los recuentos de linfocitos T circulantes y subconjuntos de linfocitos T (CD3+, CD4+ y CD8+) fueron pequeños e inconsistentes. Se desconoce el significado clínico de estos cambios. Los niveles totales de IgG, IgM e IgA en suero después de 6 meses de administración en pacientes con artritis reumatoidea fueron menores que el placebo; sin embargo, los cambios fueron pequeños y no dependientes de la dosis. Después del tratamiento con Tofacitinib en pacientes con artritis reumatoidea se observaron disminuciones rápidas de la proteína C reactiva (PCR) en suero y se mantuvieron a lo largo del tratamiento. Los cambios observados en la PCR con el tratamiento con Tofacitinib no revierten completamente dentro de las 2 semanas posteriores a la suspensión, lo que indica una mayor duración de la actividad farmacodinámica en comparación con la vida media farmacocinética. Se han observado cambios similares en las células T, las células B y la PCR sérica en pacientes con artritis psoriásica activa, aunque no se evaluó la reversibilidad. No se evaluaron las inmunoglobulinas séricas totales en pacientes con artritis psoriásica activa. Farmacocinética: Tras la administración oral de Tofacitinib, las concentraciones plasmáticas máximas se alcanzan dentro de 0,5-1 hora, la vida media de eliminación es de aproximadamente 3 horas y, dentro del rango terapéutico, se observó un aumento en la exposición sistémica proporcional a la dosis. Se obtienen concentraciones en estado estacionario en 24-48 horas, con una acumulación insignificante después de la administración dos veces al día. Absorción: La biodisponibilidad oral absoluta de Tofacitinib es del 74%. La administración concomitante de Tofacitinib con una comida de alto contenido graso no produjo ningún cambio en el área bajo la curva (ABC) mientras que la concentración plasmática máxima (Cmáx) se vio reducida en un 32%. En estudios clínicos, Tofacitinib se administró independientemente de las comidas. Distribución: Después de la administración intravenosa, el volumen de distribución es de 87 litros. La unión a las proteínas plasmáticas es de aproximadamente 40%. Tofacitinib se une principalmente a la albúmina y no parece unirse a la alfa-1 glicoproteína ácida. Tofacitinib se distribuye igualmente entre los glóbulos rojos y el plasma. Metabolismo y eliminación: Los mecanismos de depuración de Tofacitinib corresponden aproximadamente 70% al metabolismo hepático y 30% a la excreción renal de droga madre. El metabolismo de Tofacitinib es mediado principalmente por CYP3A4 con menor contribución de CYP2C19. En un estudio realizado en humanos con el fármaco radiomarcado, más del 65% del total de radiactividad circulante correspondió a Tofacitinib sin cambios y el 35% restante se atribuyó a 8 metabolitos, representando cada uno menos del 8% de la radiactividad total. La actividad farmacológica de Tofacitinib se atribuye a la molécula madre. Farmacocinética en poblaciones de pacientes: Los análisis farmacocinéticos poblacionales indicaron que las características farmacocinéticas eran similares entre los pacientes con artritis reumatoidea, artritis psoriásica y CU. El coeficiente de variación (%) en el ABC de Tofacitinib fue generalmente similar en pacientes con diferentes enfermedades, oscilando entre el 22% y el 34%. Poblaciones específicas: El análisis de farmacocinética poblacional realizado en pacientes no indicó ningún cambio clínicamente relevante en la exposición a Tofacitinib, luego de tener en cuenta las diferencias en la función renal (clearance de creatinina) entre pacientes, basado en la edad, peso, sexo y raza. Se observó una relación aproximadamente lineal entre el peso corporal y el volumen de distribución, dando como resultado un mayor pico (concentración plasmática máxima - Cmáx) y concentraciones valle (concentración plasmática mínima -Cmín) más bajas en los pacientes menos pesados. Sin embargo, esta diferencia no se considera clínicamente relevante. En sujetos con insuficiencia renal en etapa terminal mantenidos en hemodiálisis, el ABC media fue aproximadamente un 40% mayor en comparación con los datos históricos de sujetos sanos, lo que coincide con una contribución de aproximadamente el 30% del clearance renal al clearance total de Tofacitinib. Se recomienda el ajuste de dosis en pacientes con insuficiencia renal en etapa terminal mantenidos en hemodiálisis. Interacciones medicamentosas: Potencial de Tolvar para influir sobre la farmacocinética de otros fármacos: Los estudios in vitro indican que Tofacitinib no inhibe ni induce significativamente la actividad de las principales CYP metabolizadoras de fármacos en humanos (CYP1A2, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6 y CYP3A4) en concentraciones superiores a 160 veces la Cmáx en estado estacionario de una dosis de 10 mg dos veces al día. Estos resultados in vitro fueron confirmados por un estudio de interacción medicamentosa en humanos que demostró la inexistencia de cambios en la farmacocinética de midazolam, un sustrato de CYP3A4 altamente sensible, cuando se administró concomitantemente con Tofacitinib. Estudios in vitro indican que Tofacitinib no inhibe significativamente la actividad de las principales 5'-difosfo-glucuronosiltransferasas (UGT) de la uridina metabolizadora de fármacos en humanos (UGT1A1, UGT1A4, UGT1A6, UGT1A9 y UGT2B7), en concentraciones que exceden 250 veces la Cmáx en estado estacionario, de una dosis de 10 mg dos veces al al día. En pacientes con artritis reumatoidea, la depuración oral de Tofacitinib no varía con el tiempo, indicando que Tofacitinib no normaliza la actividad de las enzimas CYP en pacientes con artritis reumatoidea. Por lo tanto, la administración concomitante con Tolvar no debiera causar aumentos clínicamente relevantes en el metabolismo de los sustratos de las enzimas CYP en pacientes con artritis reumatoidea. Los datos in vitro indican que es baja la posibilidad de que Tofacitinib inhiba los transportadores tales como P-glicoproteina, transportadores aniónicos o catiónicos orgánicos en concentraciones terapéuticas. Potencial de otros fármacos para influir sobre la farmacocinética de Tolvar: Debido a que Tofacitinib se metaboliza por CYP3A4, es probable la interacción con fármacos que inhiben o inducen CYP3A4. Es improbable que los inhibidores de CYP2C19 como único agente o P-glicoproteína alteren sustancialmente la farmacocinética de Tofacitinib. Toxicología no clínica: Carcinogénesis, mutagénesis, trastornos de fertilidad: En un estudio toxicológico de 39 semanas en monos, Tofacitinib a niveles de exposición aproximadamente 6 veces la dosis recomendada de 5 mg dos veces al día y aproximadamente 3 veces la dosis de 10 mg dos veces al día (en base al ABC a dosis orales de 5 mg/kg dos veces al día diariamente) produjeron linfomas. No se observaron linfomas en este estudio a niveles de exposición de 1 vez la dosis recomendada de 5 mg dos veces al día y aproximadamente 0,5 veces la dosis de 10 mg dos veces al día (en base al ABC a dosis orales de 1 mg/kg dos veces al día). El potencial carcinogénico de Tofacitinib se evaluó en estudios de carcinogenicidad en ratones transgénicos rasH2 de 6 meses y en estudios de carcinogenicidad en ratas de 2 años. Tofacitinib, a niveles de exposición aproximadamente 34 veces la dosis recomendada de 5 mg dos veces al día y aproximadamente 17 veces la dosis de 10 mg dos veces al día (en base al ABC a dosis orales de 200 mg/kg/día) no fue carcinogénico en ratones. En el estudio de carcinogenicidad oral de 24 meses en ratas Sprague-Dawley, Tofacitinib causó tumores benignos de células de Leydig, hibernomas (malignidad del tejido adiposo pardo) y timomas benignos a dosis mayores o iguales a 30 mg / kg / día (aproximadamente 42 veces los niveles de exposición a la dosis recomendada de 5 mg dos veces al día, y aproximadamente 21 veces la dosis de 10 mg dos veces al día en base al ABC). Se desconoce la relevancia de los tumores benignos de células de Leydig para el riesgo humano. Tofacitinib no fue mutagénico en el ensayo de mutación inversa bacteriana. Fue positivo para clastogenicidad en el ensayo de aberración cromosómica in vitro con linfocitos humanos en presencia de enzimas metabólicas, pero negativo en ausencia de enzimas metabólicas. Tofacitinib fue negativo en el ensayo de micronúcleos de rata in vivo y en el ensayo de CHO-HGPRT in vitro y en el ensayo de síntesis de ADN no programado de hepatocitos de rata in vivo. En ratas, Tofacitinib a niveles de exposición aproximadamente 17 veces la dosis recomendada de 5 mg dos veces al día, y aproximadamente 8,3 veces la dosis de 10 mg dos veces al día (en base al ABC a dosis orales de 10 mg/kg/día) redujo la fertilidad femenina debido a aumento de la pérdida post-implantación. No hubo deterioro de la fertilidad de las ratas hembras a niveles de exposición de Tofacitinib iguales a la dosis recomendada de 5 mg dos veces al día y aproximadamente 0,5 veces la dosis de 10 mg dos veces al día (en base al ABC a dosis orales de 1 mg/kg/día). Los niveles de exposición a Tofacitinib de aproximadamente 133 veces la dosis recomendada de 5 mg dos veces al día y aproximadamente 67 veces la dosis de 10 mg dos veces al día (en base al ABC a dosis orales de 100 mg/kg/día) no tuvieron efecto sobre la fertilidad masculina, esperma motilidad o concentración de esperma.
Indicaciones.
Artritis reumatoidea: Tratamiento de pacientes adultos con artritis reumatoidea (AR) activa de moderada a grave que han tenido una respuesta inadecuada o intolerancia al metotrexato. Puede usarse como monoterapia o en combinación con metotrexato u otros fármacos antirreumáticos modificadores de la enfermedad (DMARD según su sigla en idioma inglés) no biológicos. Limitaciones de uso: no se recomienda el uso de Tolvar en combinación con DMARD biológicos o con inmunosupresores potentes como azatioprina y ciclosporina. Artritis psoriásica: Tratamiento de pacientes adultos con artritis psoriásica activa (APs) que han tenido una respuesta inadecuada o intolerancia al metotrexato o DMARD. Limitaciones de uso: no se recomienda el uso de Tolvar en combinación con DMARD biológicos o con inmunosupresores potentes como azatioprina y ciclosporina. Colitis ulcerosa: Tratamiento de pacientes adultos con colitis ulcerosa (CU) activa moderada a grave que han tenido una respuesta insuficiente, una pérdida de respuesta o han sido intolerantes al tratamiento convencional o a un medicamento biológico. Limitaciones de uso: no se recomienda el uso de Tolvar en combinación con DMARD biológicos o inmunosupresores potentes como azatioprina y ciclosporina.
Dosificación.
Instrucciones importantes de administración: No comenzar el tratamiento con Tofacitinib en pacientes con recuento absoluto de linfocitos menor a 500 células/mm3, un recuento absoluto de neutrófilos menor a 1000 células/mm3 o que tienen niveles de hemoglobina menores a 9 g/dL. Se recomienda la interrupción de la dosis para el manejo de linfopenia, neutropenia y anemia (ver Advertencias y Precauciones y Reacciones adversas). Interrumpa el uso de Tofacitinib si el paciente desarrolla una infección seria, hasta que la infección esté controlada. Tofacitinib se administra en forma oral con o sin alimentos. Artritis reumatoidea y artritis psoriásica: La tabla 1 muestra la dosis recomendada diaria en adultos de Tofacitinib y los ajustes de dosis para pacientes que reciben inhibidores de CYP2C19 y/o CYP3A4 concomitantemente, pacientes con insuficiencia renal moderada a severa (incluyendo, pero no limitado a aquellos pacientes con insuficiencia severa, que están siendo sometidos a hemodiálisis) o hepática moderada a severa, con linfopenia, neutropenia o anemia.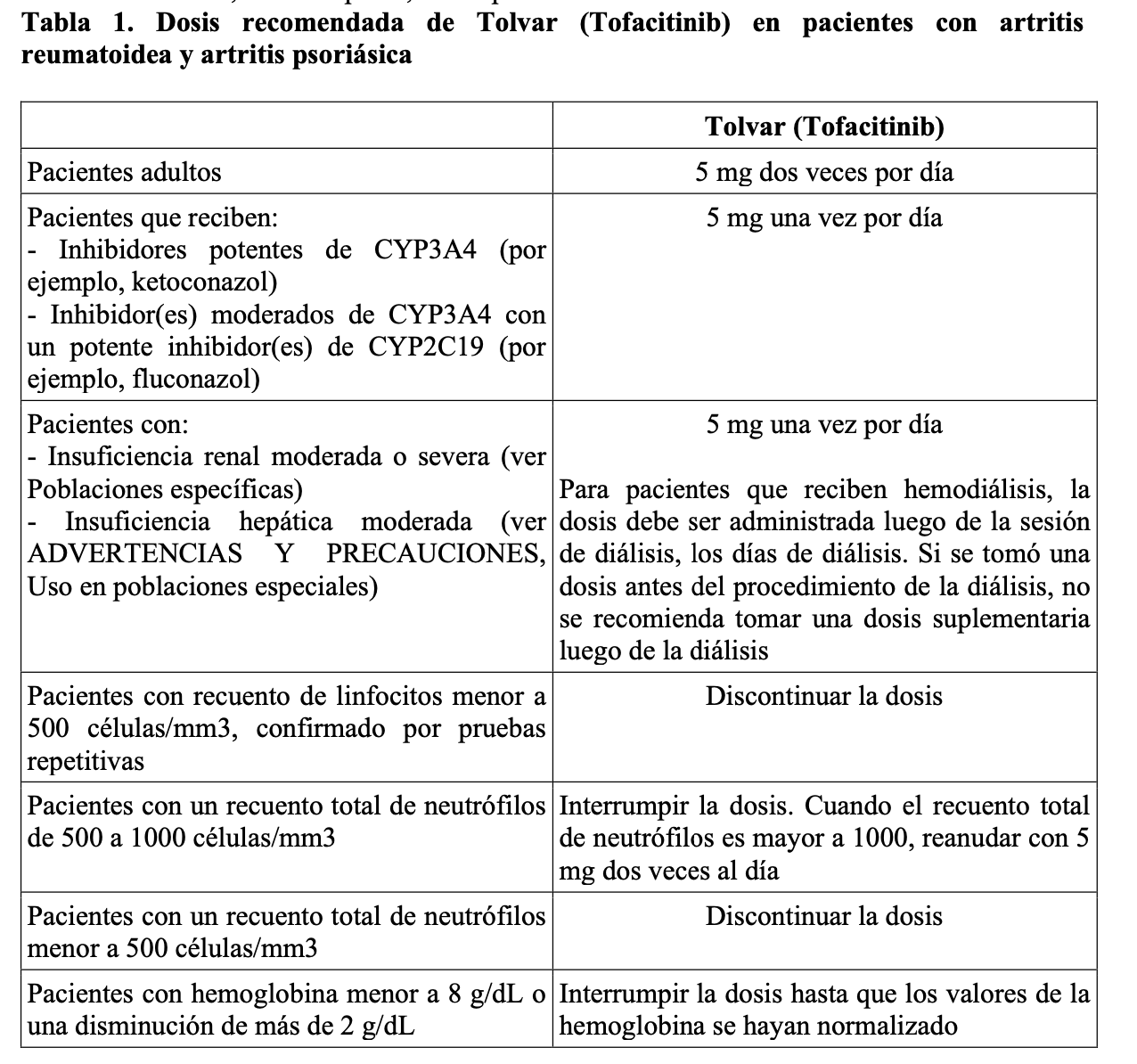
Tofacitinib puede usarse como monoterapia o en combinación con metotrexato u otros fármacos antirreumáticos no biológicos modificadores de la enfermedad para artritis reumatoidea. Tofacitinib se usa en combinación con antirreumáticos no biológicos modificadores de la enfermedad para artritis psoriásica. La eficacia de Tofacitinib como monoterapia no ha sido estudiada en artritis psoriásica. No se recomienda el uso de Tofacitinib en pacientes con insuficiencia hepática severa. Colitis ulcerosa: La dosis recomendada es de 10 mg administrados dos veces al día por vía oral para la inducción durante 8 semanas, y de 5 mg administrados dos veces al día como mantenimiento. En los pacientes que no alcancen un beneficio terapéutico adecuado en la semana 8, la dosis de inducción de 10 mg dos veces al día se puede extender durante 8 semanas adicionales (16 semanas en total), seguidas de 5 mg dos veces al día como mantenimiento. La terapia de inducción con Tofacitinib se debe suspender en los pacientes que no muestren indicios de beneficio terapéutico en la semana 16. Mantenimiento: 5 mg dos veces al día. No se recomienda Tofacitinib 10 mg dos veces al día para el tratamiento de mantenimiento en pacientes con CU con factores de riesgo conocidos de tromboembolismo venoso (TEV), a menos que no haya un tratamiento alternativo adecuado disponible. El uso de 10 mg dos veces al día más allá de la inducción debe limitarse a aquellos pacientes con pérdida de respuesta que previamente fallaron a inhibidores del TNF y usarse durante el menor tiempo posible, con una cuidadosa consideración de los beneficios y riesgos para el paciente individual. Se debe usar la dosis efectiva más baja necesaria para mantener la respuesta. En pacientes que han respondido al tratamiento con Tofacitinib, los corticosteroides se pueden reducir y/o suspender de acuerdo a la práctica clínica habitual. La Tabla 2 muestra los ajustes de dosis de Tolvar para pacientes adultos que reciben inhibidores de CYP2C19 y / o CYP3A4, con insuficiencia renal moderada o grave (incluidos, entre otros, aquellos con insuficiencia grave que se someten a hemodiálisis) o deterioro de la función hepática moderada, con linfopenia, neutropenia o anemia.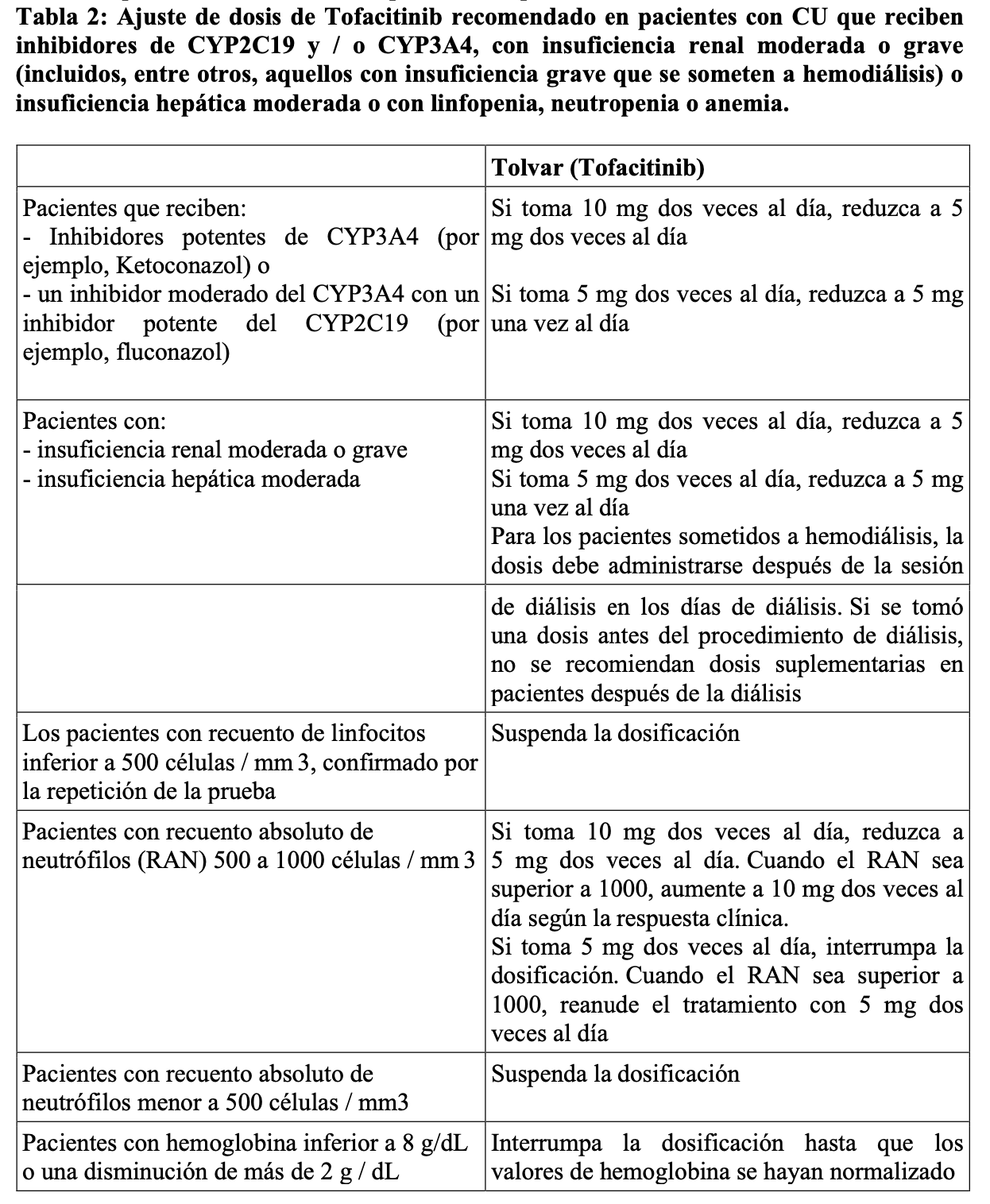
Contraindicaciones.
Pacientes con hipersensibilidad al Tofacitinib o a algún otro componente de Tolvar. Tuberculosis activa, infecciones graves y activas como sepsis o infecciones oportunistas. Insuficiencia hepática grave. Embarazo y lactancia (ver Advertencias y Precauciones).
Reacciones adversas.
Artirtis reumatoidea: Las reacciones adversas serias más frecuentes fueron infecciones serias (ver "Advertencias y Precauciones"). La proporción de pacientes que recibieron Tofacitinib durante 3 meses y debieron suspender el tratamiento por cualquier reacción adversa fue del 4% para los pacientes que recibieron Tofacitinib y del 3% para los pacientes tratados con placebo. Infecciones generales: La frecuencia de infecciones fue del 20% y 22% en los pacientes que recibieron durante 3 meses Tofacitinib 5 mg dos veces al día y 10 mg dos veces al día, respectivamente, y del 18% en el grupo placebo. Las infecciones informadas con mayor frecuencia con Tofacitinib fueron infecciones de las vías respiratorias superiores, nasofaringitis e infecciones de las vías urinarias (4%, 3% y 2% de los pacientes, respectivamente). Infecciones serias: Se observaron infecciones serias en 1 paciente (0,5 eventos por 100 pacientes-año) que recibió placebo y en 11 pacientes (1,7 eventos por 100 pacientes-año) que recibieron durante 3 meses 5 mg de Tofacitinib dos veces al día o 10 mg de Tofacitinib dos veces al día. La diferencia del índice entre los grupos de tratamiento (y el correspondiente intervalo de confianza del 95%) fue de 1,1 (-0,4, 2,5) eventos por 100 pacientes-año para el grupo combinado que recibió Tofacitinib 5 mg dos veces al día y 10 mg dos veces al día menos placebo. Se observaron infecciones serias en 34 pacientes (2,7 eventos por 100 pacientes-año) que recibieron durante 12 meses 5 mg de Tofacitinib dos veces al día y en 33 pacientes (2,7 eventos por 100 pacientes-año) que recibieron durante 12 meses 10 mg de Tofacitinib dos veces al día. La diferencia del índice entre las dosis de Tofacitinib (y el correspondiente intervalo de confianza del 95%) fue de -0,1 (-1,3, 1,2) eventos por 100 pacientes-año para 10 mg de Tofacitinib dos veces al día menos 5 mg de Tofacitinib dos veces al día. Las infecciones serias que se informaron con mayor frecuencia con el uso de Tofacitinib incluyeron neumonía, celulitis, herpes zoster e infección de las vías urinarias. Tuberculosis: Durante 3 meses de tratamiento con Tofacitinib, no se observó tuberculosis en los pacientes que recibieron Tofacitinib o placebo en cualquier nivel de dosis. Se informó tuberculosis en 0 pacientes que recibieron 5 mg de Tofacitinib dos veces al día y en 6 pacientes (0,5 eventos por 100 pacientes-año) que recibieron 10 mg de Tofacitinib dos veces al día tratados en ambos casos durante 12 meses. La diferencia del índice entre las dosis de Tofacitinib (y el correspondiente intervalo de confianza del 95%) fue de 0,5 (0,1, 0,9) eventos por 100 pacientes-año para 10 mg de Tofacitinib dos veces al día menos 5 mg Tofacitinib dos veces al día. También se observaron casos de tuberculosis diseminada. La mediana de la exposición a Tofacitinib antes del diagnóstico de tuberculosis fue de 10 meses (rango de 152 a 960 días). Infecciones oportunistas (excluida tuberculosis): Al considerarse 3 meses de tratamiento, no se observaron infecciones oportunistas en los pacientes que recibieron Tofacitinib o placebo. Al considerarse 12 meses de tratamiento, se evidenciaron infecciones oportunistas en 4 pacientes (0,3 eventos por 100 pacientes-año) que recibieron 5 mg de Tofacitinib dos veces al día y en 4 pacientes (0,3 eventos por 100 pacientes-año) que recibieron 10 mg de Tofacitinib dos veces al día. La diferencia del índice entre las dosis de Tofacitinib (y el correspondiente intervalo de confianza del 95%) fue de 0 (-0,5, 0,5) eventos por 100 pacientes-año para 10 mg de Tofacitinib dos veces al día menos 5 mg de Tofacitinib dos veces al día. La mediana de la exposición a Tofacitinib antes del diagnóstico de una infección oportunista fue de 8 meses (rango de 41 a 698 días) (ver "Advertencias y Precauciones"). Tumores malignos: Al considerarse 3 meses de tratamiento, se observaron tumores malignos excluido NMSC en 0 pacientes que recibieron placebo y en 2 pacientes (0,3 eventos por 100 pacientes-año) que recibieron 5 mg ó 10 mg de Tofacitinib dos veces al día. La diferencia del índice entre los grupos de tratamiento (y el correspondiente intervalo de confianza del 95%) fue de 0,3 (-0,1, 0,7) eventos por 100 pacientes-año para el grupo combinado que recibió Tofacitinib 5 mg dos veces al día y 10 mg dos veces al día menos placebo. Al considerarse 12 meses de tratamiento, se observaron tumores malignos excluido NMSC en 5 pacientes (0,4 eventos por 100 pacientes-año) que recibieron 5 mg de Tofacitinib dos veces al día y en 7 pacientes (0,6 eventos por 100 pacientes-año) que recibieron 10 mg de Tofacitinib dos veces al día, la diferencia del índice entre las dosis de Tofacitinib (y el correspondiente intervalo de confianza del 95%) fue de 0,2 (-0,4, 0,7) eventos por 100 pacientes-año para 10 mg de Tofacitinib dos veces al día menos 5 mg de Tofacitinib dos veces al día. Uno de estos tumores malignos fue un caso de linfoma que tuvo lugar durante el periodo de 0 a 12 meses en un paciente tratado con 10 mg de Tofacitinib dos veces al día. Los tipos de tumores malignos más frecuentes, incluso los tumores malignos observados durante la extensión a largo plazo, fueron cáncer de pulmón y de mama, seguido por cáncer gástrico, colorrectal, de células renales, de próstata, linfoma y melanoma maligno (ver "Advertencias y Precauciones"). Pruebas de laboratorio: Linfopenia: Se evidenciaron disminuciones en los recuentos de linfocitos por debajo de 500 células/mm3 en 0,04% de los pacientes de los grupos combinados que recibieron 5 mg de Tofacitinib dos veces al día y 10 mg de Tofacitinib dos veces al día durante los primeros 3 meses de exposición. Los recuentos confirmados de linfocitos inferiores a 500 células/mm3 se asociaron con un aumento en la incidencia de infecciones tratadas y serias (ver "Advertencias y Precauciones"). Neutropenia: Se observaron disminuciones confirmadas en el recuento absoluto de neutrófilos por debajo de 1000 células/mm3, tuvieron lugar en 0,07% de los pacientes de los grupos combinados que recibieron 5 mg de Tofacitinib dos veces al día y 10 mg Tofacitinib dos veces al día durante los primeros 3 meses de exposición. No se observaron disminuciones confirmadas en el recuento absoluto de neutrófilos por debajo de 500 células/mm3 en ningún grupo de tratamiento. No hubo una relación clara entre neutropenia y la aparición de infecciones serias. En los pacientes tratados a largo plazo, el patrón y la incidencia de disminuciones confirmadas en el recuento absoluto de neutrófilos fueron similares (ver "Advertencias y Precauciones"). Pruebas de enzimas hepáticas: Se observaron aumentos confirmados en las enzimas hepáticas superiores a 3 veces el límite superior de lo normal (3 x LSN) en los pacientes tratados con Tofacitinib. En los pacientes que tuvieron una elevación de las enzimas hepáticas, la modificación de la pauta posológica tal como la reducción de la dosis del fármaco antirreumático modificador de la enfermedad concomitante, suspensión de Tofacitinib o reducción de la dosis de Tofacitinib, produjo una disminución o normalización de las enzimas hepáticas. Con Tofacitinib como monoterapia durante 3 meses, no se observaron diferencias en la incidencia de elevaciones de alanina aminotransferasa (ALT) o aspartato aminotransferasa (AST) entre los grupos placebo y los que recibieron 5 mg y 10 mg de Tofacitinib dos veces al día. Con Tofacitinib asociado con drogas modificadores de la artritis reumatoidea durante 3 meses, se observaron elevaciones de ALT superiores a 3 x LSN en el 1,0%, 1,3% y 1,2% de los pacientes que recibieron placebo, 5 mg y 10 mg dos veces al día, respectivamente. Además, se observaron elevaciones de AST superiores a 3 x LSN en el 0,6%, 0,5% y 0,4% de los pacientes que recibieron placebo, 5 mg y 10 mg dos veces al día, respectivamente. Se informó un caso de lesión hepática inducida por el fármaco en un paciente tratado con 10 mg de Tofacitinib dos veces al día durante aproximadamente 2,5 meses. El paciente presentó elevaciones sintomáticas de AST y ALT superiores a 3 x LSN y elevaciones de los niveles de bilirrubina superiores a 2 x LSN que requirieron hospitalizaciones y una biopsia hepática. Lípidos: Se observaron elevaciones relacionadas con la dosis en los parámetros lipídicos (colesterol total, colesterol LDL, colesterol HDL, triglicéridos) a un mes de exposición y permanecieron estables en adelante. A continuación, se resumen los cambios en los parámetros lipídicos durante los primeros 3 meses de exposición: La media del colesterol LDL aumentó un 15% en el grupo que recibió 5 mg de Tofacitinib dos veces al día y un 19% en el grupo que recibió 10 mg de Tofacitinib dos veces al día. La media del colesterol HDL aumentó un 10% en el grupo que recibió 5 mg de Tofacitinib dos veces al día y un 12% en el grupo que recibió 10 mg de Tofacitinib dos veces al día. La media de los cocientes de LDL/HDL permanecieron esencialmente sin cambios en los pacientes tratados con Tofacitinib. Las elevaciones en el colesterol LDL y ApoB disminuyeron a los niveles previos al tratamiento en respuesta al tratamiento con estatina. En la población de seguridad a largo plazo, las elevaciones en los parámetros lipídicos fueron similares. Creatinina sérica: Se observaron elevaciones relacionadas con la dosis en la creatinina sérica con el tratamiento con Tofacitinib. La media del aumento en la creatinina sérica fue < 0,1 mg/dl a los 12 meses de tratamiento. Sin embargo, con el aumento en la duración de la exposición en las extensiones a largo plazo, hasta el 2% de los pacientes suspendieron el tratamiento con Tofacitinib debido a un aumento en la creatinina de más del 50% con respecto al valor inicial. Se desconoce el significado clínico de las elevaciones de creatinina sérica observadas. Otras reacciones adversas: En la tabla siguiente se resumen las reacciones adversas que ocurrieron en el 2% o más de los pacientes que recibieron 5 mg o 10 mg de Tofacitinib dos veces al día y en al menos un 1% más que lo observado en los pacientes que recibieron placebo con o sin fármacos antirreumáticos modificadores de la enfermedad.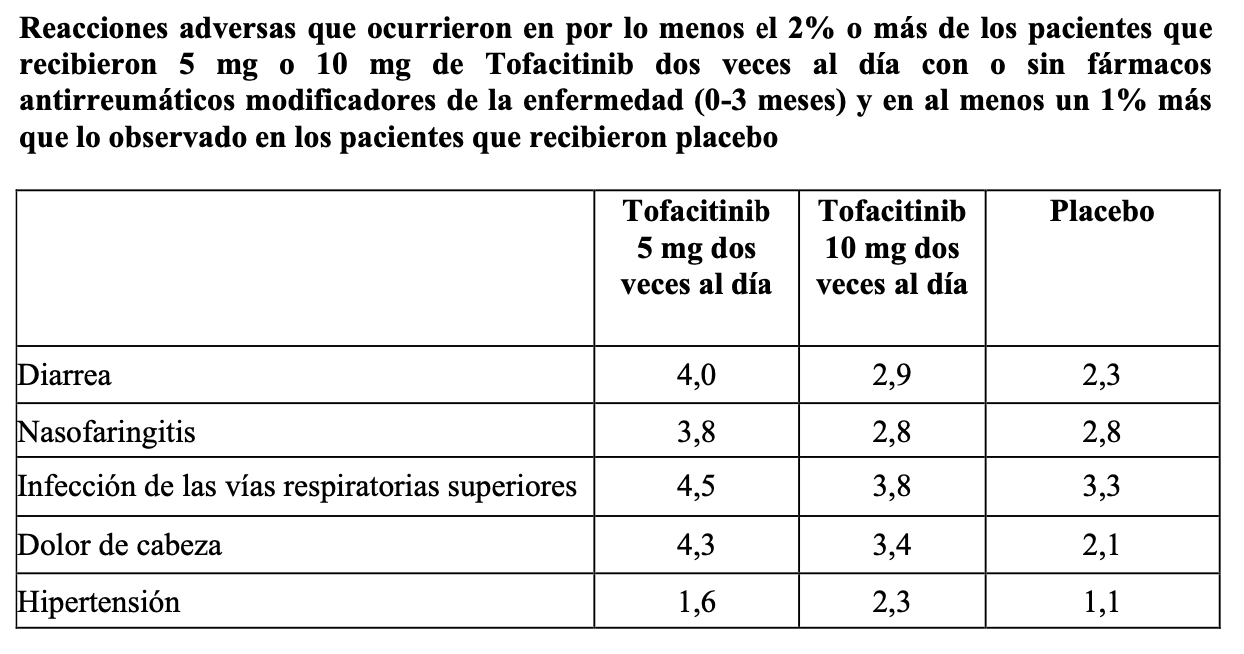
A continuación, se indican otras reacciones adversas: Trastornos de la sangre y del sistema linfático: anemia. Infecciones e infestaciones: diverticulitis. Trastornos del metabolismo y de la nutrición: deshidratación. Trastornos psiquiátricos: insomnio. Trastornos del sistema nervioso: parestesia. Trastornos respiratorios, torácicos y mediastínicos: disnea, tos, congestión sinusal, enfermedad pulmonar intersticial (en algunos casos fatales). Trastornos gastrointestinales: dolor abdominal, dispepsia, vómitos, gastritis, náuseas. Trastornos hepatobiliares: esteatosis hepática. Trastornos de la piel y del tejido subcutáneo: erupción cutánea, eritema, prurito. Trastornos musculoesqueléticos y del tejido conjuntivo: dolor musculoesquelético, artralgia, tendinitis, inflamación articular. Neoplasias benignas, malignas o inespecíficas (incluyendo quistes y pólipos): cáncer de piel no melanoma. Trastornos generales y alteraciones en el lugar de administración: pirexia, fatiga, edema periférico. Artritis psoriásica: Tofacitinib 5 mg dos veces al día y 10 mg dos veces al día se estudiaron en 2 ensayos clínicos de fase 3 doble ciego en pacientes con APs activa. Aunque se han estudiado otras dosis de Tofacitinib, la dosis recomendada de Tofacitinib es de 5 mg dos veces al día. No se recomienda una dosis de Tofacitinib 10 mg dos veces al día para el tratamiento de la APs. En dos ensayos clínicos combinados de fase 3, los pacientes fueron aleatorizados y tratados con Tofacitinib 5 mg dos veces al día o 10 mg dos veces al día. Todos los pacientes de los ensayos clínicos debían recibir tratamiento con una dosis estable de un DMARD no biológico (la mayoría -79%- recibió metotrexato). La población de estudio aleatorizada y tratada con Tofacitinib incluyó a 45 (9,5%) pacientes de 65 años o más y 66 (13,9%) pacientes con diabetes al inicio del estudio. El perfil de seguridad observado en pacientes con APs activa tratados con Tofacitinib fue consistente con el perfil de seguridad observado en pacientes con artritis reumatoidea. Colitis ulcerosa: se ha estudiado en pacientes con CU activa de moderada a grave en 4 ensayos aleatorizados, doble ciego, controlados con placebo y un ensayo abierto de extensión a largo plazo. Las reacciones adversas notificadas en ≥5% de los pacientes tratados con 5 mg o 10 mg dos veces al día de Tofacitinib y ≥1% más que las notificadas en pacientes que recibieron placebo en los ensayos clínicos de inducción o mantenimiento fueron: nasofaringitis, niveles elevados de colesterol, cefalea, infección del tracto respiratorio superior, aumento de la creatinfosfoquinasa en sangre, erupción cutánea, diarrea y herpes zoster. Ensayos de inducción: Las reacciones adversas frecuentes notificadas en ≥2% de los pacientes tratados con Tofacitinib 10 mg dos veces al día y ≥1% mayores que las notificadas en pacientes que recibieron placebo en los ensayos de inducción fueron: dolor de cabeza, nasofaringitis, niveles elevados de colesterol, acné, aumento de la creatinfosfoquinasa en sangre y pirexia. Ensayos de mantenimiento: Las reacciones adversas comunes notificadas en ≥4% de los pacientes tratados con cualquiera de las dosis de Tofacitinib y ≥1% mayores que las notificadas en pacientes que recibieron placebo fueron: nasofaringitis, niveles de colesterol elevados, dolor de cabeza, infección del tracto respiratorio superior, aumento de la creatinfosfoquinasa en sangre, erupción, diarrea, infección por herpes, gastroenteritis, anemia, náusea. En el estudio de extensión a largo plazo, se observaron neoplasias malignas (incluidos cánceres sólidos, linfomas y CPNM) con más frecuencia en pacientes tratados con Tofacitinib 10 mg dos veces al día. Se informaron cuatro casos de embolia pulmonar en pacientes que tomaban Tofacitinib 10 mg dos veces al día, incluida una muerte en un paciente con cáncer avanzado. Las reacciones adversas dependientes de las dosis, observadas en pacientes tratados con Tofacitinib 10 mg dos veces al día, en comparación con 5 mg dos veces al día, incluyen las siguientes: infecciones por herpes zoster, infecciones graves y CPNM. Experiencia posterior a la comercialización: Se han identificado las siguientes reacciones adversas durante el uso posterior a la aprobación de Tofacitinib. Debido a que estas reacciones se informan voluntariamente de una población de tamaño incierto, no siempre es posible estimar de manera confiable su frecuencia o establecer una relación causal con la exposición al fármaco. Trastornos del sistema inmunológico: hipersensibilidad al fármaco (se han observado acontecimientos como angioedema y urticaria).
Advertencias.
Infecciones graves: Los pacientes tratados con Tofacitinib tienen un mayor riesgo de desarrollar infecciones graves que pueden llevar a la hospitalización o la muerte. La mayoría de los pacientes que desarrollaron estas infecciones estaban tomando inmunosupresores concomitantes como metotrexato o corticosteroides. Si se desarrolla una infección grave, interrumpa Tofacitinib hasta que la infección esté controlada. Las infecciones reportadas incluyen: Tuberculosis activa, que puede presentarse con enfermedad pulmonar o extrapulmonar. Los pacientes deben someterse a pruebas de tuberculosis latente antes del uso de Tofacitinib y durante la terapia. El tratamiento para la infección latente debe iniciarse antes del uso de Tofacitinib. Infecciones fúngicas invasivas, que incluyen criptococosis y neumocistosis. Los pacientes con infecciones fúngicas invasivas pueden presentar una enfermedad diseminada, en lugar de localizada. Infecciones bacterianas, virales, incluido el herpes zoster y otras debidas a patógenos oportunistas. Los riesgos y beneficios del tratamiento con Tofacitinib deben considerarse cuidadosamente antes de iniciar el tratamiento en pacientes con infección crónica o recurrente. Se debe vigilar estrechamente a los pacientes para detectar el desarrollo de signos y síntomas de infección durante y después del tratamiento con Tofacitinib, incluido el posible desarrollo de tuberculosis en pacientes que resultaron negativos para la infección tuberculosa latente antes de iniciar el tratamiento. Mortalidad: Los pacientes con artritis reumatoidea de 50 años o más con al menos un factor de riesgo cardiovascular (CV) tratados con Tofacitinib 10 mg dos veces al día tuvieron una tasa más alta de mortalidad por todas las causas, incluida la muerte súbita CV, en comparación con los tratados con Tofacitinib 5 mg administrados dos veces al día o inhibidores del TNF en un gran estudio de seguridad posterior a la comercialización en curso. Tumores malignos: Se han observado linfomas y otras neoplasias malignas en pacientes tratados con Tofacitinib. El trastorno linfoproliferativo postrasplante asociado al virus de Epstein Barr se ha observado a una tasa mayor en pacientes con trasplante renal tratados con Tofacitinib y medicamentos inmunosupresores concomitantes. Trombosis: Se han producido trombosis, incluida embolia pulmonar, trombosis venosa profunda y trombosis arterial en pacientes tratados con Tofacitinib y otros inhibidores de la cinasa Janus utilizados para tratar enfermedades inflamatorias. Los pacientes con artritis reumatoidea que tenían 50 años de edad o más con al menos un factor de riesgo CV tratados con Tofacitinib 10 mg dos veces al día en comparación con Tofacitinib 5 mg dos veces al día o inhibidores del TNF en un gran estudio de seguridad poscomercialización en curso tuvieron un aumento observado en la incidencia de estos eventos. Muchos de estos eventos fueron graves y algunos resultaron en muerte. Evite Tofacitinib en pacientes de riesgo. Suspenda el tratamiento y evalúe de inmediato a los pacientes con síntomas de trombosis. Para pacientes con colitis ulcerosa, use Tofacitinib a la dosis efectiva más baja y durante el menor tiempo necesario para lograr / mantener la respuesta terapéutica. Infecciones serias: Se han informado infecciones serias, y a veces mortales, debido a patógenos bacterianos, micobacterianos, fúngicos invasivos, virales u otros oportunistas en pacientes con artritis reumatoidea que recibieron Tofacitinib. Las infecciones serias que se informaron con mayor frecuencia con el uso de Tofacitinib incluyeron neumonía, celulitis, herpes zoster, infección de las vías urinarias, diverticulitis y apendicitis. Con el uso de Tofacitinib se han informado infecciones oportunistas como tuberculosis (que puede presentarse como enfermedad pulmonar o extrapulmonar) y otras infecciones micobacterianas, criptococosis, candidiasis esofágica, neumocistosis, herpes zoster multidermatómico, infección por citomegalovirus, virus BK y listeriosis. Algunos pacientes presentaron enfermedad diseminada en lugar de localizada y generalmente recibían agentes inmunomoduladores concomitantes tales como metotrexato o corticosteroides. También pueden ocurrir otras infecciones serias como histoplasmosis y coccidiodomicosis. No se debe comenzar el tratamiento con Tolvar en pacientes con una infección seria activa, incluso infecciones localizadas. Se deben considerar los riesgos y beneficios del tratamiento antes de comenzarlo en pacientes que presenten lo siguiente: infección crónica o recurrente, hayan sido expuestos a tuberculosis, antecedentes de una infección seria u oportunista, hayan residido o viajado en zonas de tuberculosis endémica o micosis endémica, afecciones subyacentes que puedan predisponerlos a infección. mayores de 65 años. Se debe controlar de cerca a los pacientes por la aparición de signos y síntomas de infección durante y después del tratamiento con Tolvar. Se debe suspender el tratamiento con Tolvar si el paciente presenta una infección seria, oportunista o sepsis, hasta que la infección sea controlada. Los pacientes que presenten una infección nueva durante el tratamiento con Tolvar deben ser sometidos a una prueba diagnóstica rápida y completa adecuada para pacientes inmunocomprometidos, se debe comenzar una terapia antimicrobiana apropiada y estricto control. Se recomienda también precaución en pacientes que tengan antecedentes de enfermedad pulmonar crónica, o en aquellos que presenten enfermedad pulmonar intersticial, ya que pueden ser más propensos a presentar infecciones. El riesgo de contraer infecciones puede ser mayor con grados crecientes de linfopenia. Al evaluar el riesgo individual de presentar una infección se debe considerar el recuento de linfocitos del paciente. Tuberculosis: Se debe investigar en los pacientes la presencia de infección latente o activa antes y durante la administración de Tolvar, según describen las guías clínicas. También se debe considerar la terapia antimicobacteriana antes de la administración de Tolvar en pacientes con antecedentes de tuberculosis latente o activa en quienes no se pueda confirmar un ciclo adecuado de tratamiento y en pacientes con un resultado negativo para la prueba de tuberculosis latente pero que poseen factores de riesgo para desarrollar infección por tuberculosis. Se recomienda consultar un médico con experiencia en el tratamiento de tuberculosis para que ayude a tomar la decisión sobre la conveniencia de comenzar una terapia antituberculosis en un paciente en particular. Se debe controlar estrechamente a los pacientes en busca de la aparición de signos y síntomas de tuberculosis, incluso a los pacientes cuyo resultado de la prueba por infección de tuberculosis latente fuera negativo antes de comenzar el tratamiento. Los pacientes con tuberculosis latente deben ser tratados con terapia antimicobacteriana estándar antes de la administración de Tolvar. Reactivación viral: Se observó reactivación viral, incluso casos de reactivación del virus del herpes (por ejemplo, herpes zoster), en pacientes tratados con Tofacitinib. Se desconoce el impacto de Tofacitinib sobre la reactivación de la hepatitis viral crónica. No hay datos respecto del uso de Tofacitinib en pacientes con resultados positivos para hepatitis B o C. Antes de iniciar la terapia con Tolvar se deberá realizar detección de infección por virus de la hepatitis de acuerdo a las guías clínicas. El riesgo de infección por el virus herpes zoster aumenta en pacientes tratados con Tofacitinib y sería mayor en pacientes tratados con Tofacitinib en Japón y Corea. Mortalidad: Los pacientes con artritis reumatoidea de 50 años o más con al menos un factor de riesgo cardiovascular (CV) tratados con Tofacitinib 10 mg dos veces al día tuvieron una tasa más alta de mortalidad por todas las causas, incluida la muerte súbita CV, en comparación con los tratados con Tofacitinib 5 mg. administrados dos veces al día o inhibidores del TNF en un gran estudio de seguridad posterior a la comercialización en curso. No se recomienda una dosis de Tofacitinib 10 mg dos veces al día para el tratamiento de la AR o la PsA. Para el tratamiento de la CU, se recomienda utilizar Tofacitinib a la dosis efectiva más baja y durante el menor tiempo necesario para lograr / mantener la respuesta terapéutica. Tumores malignos y trastorno linfoproliferativo: Se han observado linfo