VEMLIDY®
GADOR
Grupo farmacoterapéutico: Antivirales de uso sistémico, nucleósidos y nucleótidos inhibidores de transcriptasa reversa; código ATC: J05AF13.
Composición.
Cada comprimido recubierto de VEMLIDY® contiene: Tenofovir alafenamida (equivalente a 28,04 mg de tenofovir alafenamida fumarato) 25 mg. Excipientes: Lactosa monohidrato, Celulosa microcristalina, Croscaramelosa sódica, Estearato de magnesio, Alcohol polivinílico*, Dióxido de titanio*, Polietilenglicol*, Talco*, Óxido de hierro amarillo CI N°77492* c.s. * Se refiere a los componentes del Opadry II amarillo 85F120028. VEMLIDY® contiene lactosa monohidrato.
Farmacología.
Descripción: Comprimido recubierto. Los comprimidos de VEMLIDY® recubiertos con película son amarillos, redondos, con "GSI" impreso (o marcado) en una cara del comprimido y "25" en la otra cara del comprimido. Farmacología Clínica: Mecanismo de acción: Tenofovir alafenamida es un profármaco fosfonamidato de tenofovir (análogo de 2' desoxiadenosina monofosfato). Tenofovir alafenamida entra en los hepatocitos primarios por difusión pasiva y mediante los transportadores de captación hepática OATP1B1 y OATP1B3. Tenofovir alafenamida se hidroliza primeramente para formar tenofovir por la carboxilesterasa 1 en los hepatocitos primarios. El tenofovir intracelular se fosforila posteriormente al metabolito farmacológicamente activo, tenofovir difosfato. Tenofovir difosfato inhibe la replicación del VHB mediante su incorporación al ADN viral por la transcriptasa inversa del VHB, lo que produce la terminación de la cadena de ADN. El tenofovir tiene una actividad que es específica para el virus de la hepatitis B y el virus de la inmunodeficiencia humana (VIH-1 y VIH-2). El tenofovir difosfato es un inhibidor débil de las ADN polimerasas de mamíferos que incluyen la ADN polimerasa c mitocondrial y no hay evidencia de toxicidad mitocondrial in vitro de acuerdo a varios ensayos que incluyen análisis del ADN mitocondrial. Propiedades farmacocinéticas: Absorción: Después de la administración oral de VEMLIDY® en ayunas en pacientes adultos con hepatitis B crónica, las concentraciones plasmáticas máximas de tenofovir alafenamida se observaron aproximadamente 0,48 horas después de la dosis. De acuerdo a los análisis farmacocinéticos en sujetos con hepatitis B crónica en la población de fase III, las medias en estado estacionario de AUC0-24 de tenofovir alafenamida (N = 698) y tenofovir (N = 856) fueron de 0,22 mg•h/ml y 0,32 mg•h/ml, respectivamente. Las Cmáx. En estado estacionario de tenofovir alafenamida y tenofovir fueron de 0,18 y 0,mg/ml, respectivamente. Con relación a las condiciones de ayuno, la administración de una dosis única de VEMLIDY® con una comida de alto contenido graso resultó en un aumento del 65 % en la exposición a tenofovir alafenamida. Distribución: La unión de tenofovir alafenamida a las proteínas plasmáticas humanas en las muestras recogidas durante los estudios clínicos fue de aproximadamente el 80 %. La unión de tenofovir a las proteínas plasmáticas humanas es inferior al 0,7 % y es independiente de la concentración en el intervalo de 0,01-25 mg/ml. Biotransformación: El metabolismo es la ruta de eliminación principal de tenofovir alafenamida en los seres humanos, suponiendo > 80 % de una dosis oral. Los estudios in vitro han demostrado que tenofovir alafenamida se metaboliza a tenofovir (metabolito principal) por medio de la carboxilesterasa-1 en los hepatocitos; y por la catepsina A en las células mononucleares de sangre periférica y los macrófagos. In vivo, tenofovir alafenamida se hidroliza en las células para formar tenofovir (metabolito principal), que es fosforilado al metabolito activo, tenofovir difosfato. In vitro, tenofovir alafenamida no se metaboliza por CYP1A2, CYP2C8, CYP2C9, CYP2C19 o CYP2D6. Tenofovir alafenamida se metaboliza mínimamente por CYP3A4. Eliminación: La excreción renal de tenofovir alafenamida intacta es una ruta menor, con < 1 % de la dosis eliminada por la orina. Tenofovir alafenamida se elimina principalmente después de la metabolización a tenofovir. Tenofovir alafenamida y tenofovir tienen una mediana de semivida plasmática de 0,51 y 32,37 horas, respectivamente. Tenofovir se elimina del organismo a través de los riñones por filtración glomerular y secreción tubular activa. Linealidad/no linealidad: Las exposiciones a tenofovir alafenamida son proporcionales a la dosis a lo largo del intervalo de dosis de 8 a 125 mg. Farmacocinética en poblaciones especiales: Edad, sexo y origen étnico: No se han identificado diferencias clínicamente relevantes en la farmacocinética en función de la edad o el origen étnico. Las diferencias en la farmacocinética en función del sexo no se consideraron clínicamente relevantes. Insuficiencia hepática: En pacientes con insuficiencia hepática grave, las concentraciones plasmáticas totales de tenofovir alafenamida y tenofovir son inferiores a las observadas en sujetos con función hepática normal. Cuando se retiró la parte unida a proteínas, las concentraciones plasmáticas de tenofovir alafenamida no unida (libre) en la insuficiencia hepática grave y en la función hepática normal son similares. Insuficiencia renal: No se observaron diferencias clínicamente relevantes en la farmacocinética de tenofovir alafenamida o tenofovir entre sujetos sanos y pacientes con insuficiencia renal grave (ClCr estimada > 15 pero < 30 ml/min) en los estudios de tenofovir alafenamida. Población pediátrica: Se evaluó la farmacocinética de tenofovir alafenamida y tenofovir en adolescentes infectados por el VIH-1, sin tratamiento previo, que recibieron tenofovir alafenamida (10 mg) administrado con elvitegravir, cobicistat y emtricitabina en forma de comprimido de combinación en dosis fija (E/C/F/TAF; Genvoya). No se observaron diferencias clínicamente relevantes en la farmacocinética de tenofovir alafenamida o tenofovir entre los sujetos adolescentes y adultos infectados por el VIH-1. Microbiología: Actividad antiviral: La actividad antiviral de tenofovir alafenamida se evaluó en células HepG2 frente a un panel de VHB procedentes de aislados clínicos que representaban los genotipos A-H. Los valores de CE50 (concentración efectiva 50 %) para tenofovir alafenamida oscilaron entre 34,7 y 134,4 nM, con una media global de CE50 de 86,6 nM. La CC50 (concentración de citotoxicidad 50 %) en células HepG2 fue > 44.400 nM. Resistencia: En un análisis agrupado de pacientes recibiendo VEMLIDY®, se realizó un análisis de secuenciación de cepas de VHB en las dos muestras, la del momento basal y la muestra durante el tratamiento, en los pacientes que o bien presentaron un rebrote virológico (2 visitas consecutivas con ADN de VHB ≥69 UI/ml después de haber sido < 69 UI/ml, o un incremento superior o igual a 1,0 log10 ADN de VHB desde la cifra mínima), o pacientes con ADN de VHB ≥69 UI/ml en la semana 96 o el momento de discontinuación prematura en la semana 24 o tras esta. En los análisis de la semana 48 (N = 20) y la semana 96 (N = 72) no se identificaron sustituciones de aminoácidos asociadas a resistencia a VEMLIDY® en estas muestras (análisis genotípicos y fenotípicos). Resistencia cruzada: La actividad antiviral de tenofovir alafenamida se evaluó frente a un panel de cepas aisladas de VHB que contienen mutaciones frente a inhibidor nucleos(t)ídico de la transcriptasa inversa en cultivos de células HepG2. Las cepas de VHB que expresaban las mutaciones rtV173L, rtL180M y rtM204V/I, asociadas con resistencia a la lamivudina, siguieron siendo sensibles a tenofovir alafenamida (cambio < 2 veces en CE50). Las cepas de VHB que expresaban las mutaciones rtL180M, rtM204V más rtT184G, rtS202G o rtM250V, asociadas con resistencia al entecavir, siguieron siendo sensibles a tenofovir alafenamida. Las cepas de VHB que expresaban las mutaciones de resistencia rtA181T, rtA181V o rtN236T asociadas a adefovir, siguieron siendo sensibles a tenofovir alafenamida; sin embargo, la cepa de VHB que expresaba rtA181V más rtN236T mostró una sensibilidad reducida a tenofovir alafenamida (cambio de 3,7 veces en CE50). No se conoce la relevancia clínica de estas mutaciones.
Indicaciones.
VEMLIDY® está indicado para el tratamiento de la hepatitis B crónica en adultos y adolescentes (de 12 años de edad y mayores con un peso corporal de al menos 35 kg).
Dosificación.
El tratamiento debe ser iniciado por un médico con experiencia en el tratamiento de la hepatitis B crónica. Posología: Adultos y adolescentes (de 12 años de edad y mayores con un peso corporal de al menos 35 kg): un comprimido una vez al día. Interrupción del tratamiento: Se puede contemplar la interrupción del tratamiento en los siguientes casos: En pacientes AgHBe positivo sin cirrosis, el tratamiento debe administrarse durante al menos 6-12 meses después de que se confirme la seroconversión deHBe (pérdida del AgHBe y del ADN del VHB con detección de anti-HBe) o hasta la seroconversión de HBs o hasta que haya pérdida de eficacia (ver sección 4.4). Se recomienda la reevaluación periódica después de la interrupción del tratamiento para detectar una recaída virológica. En pacientes AgHBe negativo sin cirrosis, el tratamiento debe administrarse al menos hasta la seroconversión del HBs o hasta que haya evidencia de pérdida de eficacia. Cuando el tratamiento se prolongue durante más de 2 años, se recomienda una reevaluación periódica para confirmar que continuar con la terapia seleccionada es adecuado para el paciente. Dosis omitida: Si se omite una dosis y han transcurrido menos de 18 horas desde la hora habitual de administración, el paciente debe tomar VEMLIDY® lo antes posible y luego reanudar su pauta habitual de administración. Si se omite una dosis y han transcurrido más de 18 horas desde la hora habitual de administración, el paciente no debe tomar la dosis omitida sino simplemente reanudar la pauta habitual de administración. Si el paciente vomita en el plazo de 1 hora después de tomar VEMLIDY®, debe tomar otro comprimido. Si el paciente vomita más de 1 hora después de tomar VEMLIDY®, no es necesario que tome otro comprimido. Poblaciones especiales: Pacientes de edad avanzada: No se requiere ajustar la dosis de VEMLIDY® en pacientes de 65 años de edad y mayores. Insuficiencia renal: No se requiere ajustar la dosis de VEMLIDY® en adultos o adolescentes (de al menos 12 años de edad y al menos 35 kg de peso corporal) con aclaramiento de creatinina estimado (ClCr) ≥15 ml/min o en pacientes con ClCr < 15 ml/min que estén recibiendo hemodiálisis. En los días de hemodiálisis, VEMLIDY® se debe administrar tras completar el tratamiento de hemodiálisis. No pueden darse recomendaciones de dosis para pacientes con ClCr < 15 ml/min que no estén recibiendo hemodiálisis. Insuficiencia hepática: No se requiere ajustar la dosis de VEMLIDY® en pacientes con insuficiencia hepática. Población pediátrica: No se ha establecido todavía la seguridad y eficacia de VEMLIDY® en niños menores de 12 años de edad, o que pesen < 35 kg. No se dispone de datos. Forma de administración: Administración por vía oral. VEMLIDY® comprimidos recubiertos con película se debe tomar con alimentos. Formas farmacéuticas y concentraciones: Comprimido recubierto. Los comprimidos de VEMLIDY® recubiertos con película son amarillos, redondos, con "GSI" impreso (o marcado) en una cara del comprimido y "25" en la otra cara del comprimido.
Contraindicaciones.
Hipersensibilidad al principio activo o a alguno de los excipientes.
Reacciones adversas.
Resumen del perfil de seguridad: La evaluación de las reacciones adversas se basa en datos de seguridad agrupados de 2 estudios de fase III controlados en los cuales 866 pacientes infectados por VHB recibieron 25 mg de tenofovir alafenamida una vez al día en forma de doble ciego hasta la semana 96 (mediana de la duración de la exposición ciega al medicamento de estudio de 104 semanas) y de la experiencia post-comercialización. Las reacciones adversas notificadas con más frecuencia fueron cefalea (12 %), náuseas (6 %) y fatiga (6 %). Después de la semana 96, los pacientes permanecieron en su tratamiento ciego original o bien recibieron tratamiento abierto de VEMLIDY®. No se identificaron reacciones adversas adicionales a VEMLIDY® desde la semana 96 hasta la 120 en la fase de doble ciego ni en el subconjunto de sujetos que recibieron el tratamiento de VEMLIDY® abierto (ver Farmacología). Tabla de reacciones adversas: Se han identificado las siguientes reacciones adversas con tenofovir alafenamida en pacientes con hepatitis B crónica (Tabla 5). Las reacciones adversas se enumeran a continuación por clase de órganos y sistemas y frecuencia. Las frecuencias se definen del siguiente modo: muy frecuentes (≥1/10), frecuentes (≥1/100 a < 1/10), poco frecuentes (≥1/1.000 a < 1/100). (Ver Tabla 5).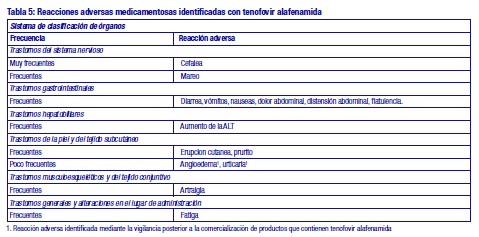
Notificación de sospechas de reacciones adversas: Es importante notificar sospechas de reacciones adversas al medicamento tras su autorización. Ello permite una supervisión continuada de la relación beneficio/riesgo del medicamento. Se invita a los profesionales sanitarios a notificar las sospechas de reacciones adversas a través del Sistema Nacional de Farmacovigilancia al siguiente link: https://www.argentina.gob.ar/anmat/farmacovigilancia/notificanos/eventosadversos y/o al Departamento de Farmacovigilancia de GADOR S.A. vía email a farmacovigilancia@gador.com o telefónicamente al 0800-220-2273.
Advertencias.
Generales: Transmisión del VHB: Se debe advertir a los pacientes que VEMLIDY® no evita el riesgo de transmisión del VHB a otras personas por contacto sexual o contaminación por sangre. Se deben seguir tomando las precauciones apropiadas. Pacientes con enfermedad hepática descompensada: No existen datos sobre la seguridad y eficacia de VEMLIDY® en pacientes infectados por el VHB con enfermedad hepática descompensada y que tienen un índice de Child Pugh Turcotte (CPT) > 9 (es decir, de clase C). Estos pacientes pueden tener un riesgo mayor de experimentar reacciones adversas hepáticas o renales graves. Por lo tanto, los parámetros hepatobiliares y renales deben ser monitorizados estrechamente en esta población de pacientes. Exacerbaciones de la hepatitis: Brotes durante el tratamiento: Las exacerbaciones espontáneas de la hepatitis B crónica son relativamente frecuentes y se caracterizan por aumentos transitorios de la alanina-aminotransferasa (ALT) sérica. Tras el inicio del tratamiento antiviral los niveles séricos de ALT sérica pueden aumentar en algunos pacientes. En pacientes con enfermedad hepática compensada, estos incrementos en la ALT sérica generalmente no van acompañados de un aumento en las concentraciones de bilirrubina sérica ni descompensación hepática. Los pacientes con cirrosis pueden tener un mayor riesgo de descompensación hepática tras la exacerbación de la hepatitis, y por tanto deben ser cuidadosamente monitorizados durante el tratamiento. Brotes después de interrumpir el tratamiento: Se ha notificado exacerbación aguda de la hepatitis en pacientes que han interrumpido el tratamiento de la hepatitis B, por lo general asociada con aumentos en los niveles de ADN del VHB en plasma. En la mayoría de los casos son autolimitadas, pero pueden ocurrir exacerbaciones graves, incluyendo muertes, después de la interrupción del tratamiento de la hepatitis B. La función hepática debe ser monitorizada a intervalos repetidos mediante seguimiento tanto clínico como de laboratorio durante al menos 6 meses tras la interrupción del tratamiento de la hepatitis B. Si es adecuado, se debe garantizar la reanudación del tratamiento de la hepatitis B. En pacientes con enfermedad hepática avanzada o cirrosis, no se recomienda interrumpir el tratamiento ya que la exacerbación de la hepatitis postratamiento puede provocar una descompensación hepática. Los brotes hepáticos son especialmente graves, y a veces mortales en pacientes con enfermedad hepática descompensada. Insuficiencia renal: Pacientes con aclaramiento de creatinina < 30 ml/min: El uso de VEMLIDY® una vez al día en pacientes con ClCr ≥15 ml/min pero < 30 ml/min y en pacientes con ClCr < 15 ml/min que estén recibiendo hemodiálisis está basado en datos farmacocinéticos muy limitados y en modelos y simulaciones. No hay datos de seguridad sobre el uso de VEMLIDY® para el tratamiento de pacientes infectados por el VHB con ClCr < 30 ml/min. No se recomienda el uso de VEMLIDY® en pacientes con ClCr < 15 ml/min que no estén recibiendo hemodiálisis. Nefrotoxicidad: No se puede descartar un posible riesgo de nefrotoxicidad resultante de la exposición crónica a niveles bajos de tenofovir debido a la administración de tenofovir alafenamida. Pacientes coinfectados por VHB y el virus de la hepatitis C o D: No existen datos sobre la seguridad y eficacia de VEMLIDY® en pacientes coinfectados por el virus de la hepatitis C o D. Se debe seguir la orientación acerca de la administración concomitante para el tratamiento de la hepatitis C. Coinfección por el virus de la hepatitis B y VIH: A todos los pacientes infectados por VHB cuyo estado de infección por VIH-1 sea desconocido se les debe ofrecer la posibilidad de que les realicen un análisis de anticuerpos de VIH antes de iniciar el tratamiento con VEMLIDY®. En pacientes coinfectados por VHB y VIH, se debe administrar VEMLIDY® de forma conjunta con otros antirretrovirales para garantizar que el paciente recibe una pauta adecuada para el tratamiento de VIH. Administración concomitante con otros medicamentos: No se debe administrar VEMLIDY® de forma conjunta con productos medicinales que contengan tenofovir alafenamida, tenofovir disoproxil fumarato o adefovir dipivoxil. No se recomienda la administración concomitante de VEMLIDY® con ciertos anticonvulsivos (por ejemplo, carbamazepina, oxcarbazepina, fenobarbital y fenitoína), antimicobacterianos (por ejemplo, rifampicina, rifabutina y rifapentina) o hierba de San Juan, los cuales son inductores de la glicoproteína P (P-gp) y pueden reducir las concentraciones plasmáticas de tenofovir alafenamida. La administración concomitante de VEMLIDY® con inhibidores potentes de la P-gp (por ejemplo, itraconazol y ketoconazol) pueden aumentar las concentraciones plasmáticas de tenofovir alafenamida. No se recomienda la administración concomitante. Intolerancia a la lactosa: VEMLIDY® contiene lactosa monohidrato. Los pacientes con intolerancia hereditaria a galactosa, insuficiencia total de lactasa o mala absorción de glucosa-galactosa, no deben tomar este medicamento. Efectos sobre la capacidad para conducir y utilizar máquinas: La influencia de VEMLIDY® sobre la capacidad para conducir y utilizar máquinas es nula o insignificante. Se debe informar a los pacientes que se ha notificado mareo durante el tratamiento con VEMLIDY®.
Interacciones.
Los estudios de interacciones se han realizado sólo en adultos. No se debe administrar VEMLIDY® de forma conjunta con medicamentos que contengan tenofovir disoproxil fumarato, tenofovir alafenamida o adefovir dipivoxil. Medicamentos que pueden afectar a tenofovir alafenamida: Tenofovir alafenamida es transportado por la P-gp y la proteína de resistencia de cáncer de mama (BCRP, por sus siglas en inglés). Se espera que los medicamentos inductores de la P-gp (por ejemplo, rifampicina, rifabutina, carbamazepina, fenobarbital o hierba de San Juan) reduzcan las concentraciones plasmáticas de tenofovir alafenamida, lo que puede ocasionar una pérdida del efecto terapéutico de VEMLIDY®. No se recomienda la administración concomitante de dichos medicamentos con VEMLIDY®. La administración concomitante de VEMLIDY® con medicamentos que inhiban la P-gp y/o la BCRP puede aumentar la concentración plasmática de tenofovir alafenamida. No se recomienda la administración concomitante de inhibidores potentes de la P-gp con VEMLIDY®. Tenofovir alafenamida es un sustrato de OATP1B1 y OATP1B3 in vitro. La distribución de tenofovir alafenamida en el organismo puede verse afectada por la actividad de OATP1B1 y/o OATP1B3. Efecto de tenofovir alafenamida sobre otros medicamentos: Tenofovir alafenamida no es un inhibidor del CYP1A2, CYP2B6, CYP2C8, CYP2C9, CYP2C19 o CYP2D6 in vitro. No es un inhibidor ni un inductor de la CYP3A in vivo. Tenofovir alafenamida no es un inhibidor de la uridina difosfato glucuronosiltransferasa (UGT) 1A1 humana in vitro. Se desconoce si tenofovir alafenamida es un inhibidor de otras enzimas UGT. La información sobre las interacciones farmacológicas de VEMLIDY® con posibles medicamentos concomitantes se resume en la Tabla 1 a continuación (el aumento está indicado como "↑", la disminución como "↓", la ausencia de cambios como "↔"; dosis única como "d.u.", dos veces al día como "d.v.d.", una vez al día como "u.v.d."; y por vía intravenosa con "IV"). Las interacciones farmacológicas descritas se basan en estudios realizados con tenofovir alafenamida, o son interacciones farmacológicas potenciales que pueden ocurrir con VEMLIDY®.
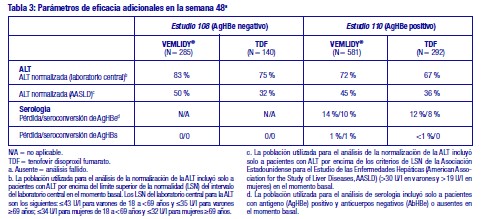
Toxicología preclínica: Los estudios no clínicos en ratas y perros mostraron los huesos y el riñón como los órganos diana primarios para la toxicidad. La toxicidad ósea fue observada en forma de reducción de la DMO en ratas y perros a unas exposiciones a tenofovir al menos cuatro veces superiores a las esperadas después de la administración de tenofovir alafenamida. Hubo una mínima infiltración de histiocitos presente en el ojo de perros con exposiciones a tenofovir alafenamida y tenofovir aproximadamente 4 y 17 veces superiores, respectivamente, a las esperadas después de la administración de tenofovir alafenamida. Tenofovir alafenamida no fue mutagénico ni clastogénico en los estudios convencionales de genotoxicidad. Dado que existe una menor exposición a tenofovir en ratas y ratones después de la administración de tenofovir alafenamida en comparación con tenofovir disoproxil fumarato, los estudios de carcinogenicidad y un estudio peri-postnatal en ratas fueron realizados solamente con tenofovir disoproxil fumarato. Los estudios convencionales de potencial carcinogénico con tenofovir disoproxil (en forma de fumarato) y toxicidad para la reproducción y el desarrollo con tenofovir disoproxil (en forma de fumarato) o tenofovir alafenamida no mostraron riesgos especiales para los seres humanos. Los estudios de toxicidad para la reproducción en ratas y conejos no mostraron ningún efecto en los parámetros de apareamiento, fertilidad y embarazo ni en ningún parámetro fetal. No obstante, tenofovir disoproxil fumarato redujo el índice de viabilidad y peso de las crías en un estudio peri-postnatal de toxicidad a dosis tóxicas para la madre. Un estudio carcinogenicidad a largo plazo por vía oral en ratones mostró una incidencia baja de tumores duodenales, que se consideraron probablemente relacionados con altas concentraciones locales en el tracto gastrointestinal a dosis altas de 600 mg/kg/día. El mecanismo de formación de tumores en ratones y su posible relevancia en seres humanos es desconocido. Uso en poblaciones específicas: Embarazo: No hay datos o estos son limitados (datos en menos de 300 embarazos) relativos al uso de tenofovir alafenamida en mujeres embarazadas. Sin embargo, existe un elevado número de datos en mujeres embarazadas (datos en más de 1.000 embarazos) que indican que el uso de tenofovir disoproxil fumarato no está asociado con malformaciones ni toxicidad fetal/neonatal. Los estudios en animales no sugieren efectos perjudiciales directos ni indirectos en términos de toxicidad para la reproducción. En casos necesarios, se puede considerar el uso de VEMLIDY® durante el embarazo. Lactancia: Se desconoce si tenofovir alafenamida se excreta en la leche materna. Sin embargo, en estudios con animales se ha demostrado que tenofovir se excreta en la leche. No hay datos suficientes sobre los efectos del tenofovir en recién nacidos/niños lactantes. No se puede excluir el riesgo en recién nacidos/niños lactantes; por lo tanto, VEMLIDY® no debe utilizarse durante la lactancia. Fertilidad: No se dispone de datos en seres humanos sobre el efecto de VEMLIDY® sobre la fertilidad. Los estudios en animales no sugieren efectos perjudiciales de tenofovir alafenamida sobre la fertilidad. Pacientes de edad avanzada: No se requiere ajustar la dosis de VEMLIDY® en pacientes de 65 años de edad y mayores (ver sección 5.2). Insuficiencia renal: No se requiere ajustar la dosis de VEMLIDY® en adultos o adolescentes (de al menos 12 años de edad y al menos 35 kg de peso corporal) con aclaramiento de creatinina estimado (ClCr) ≥15 ml/min o en pacientes con ClCr < 15 ml/min que estén recibiendo hemodiálisis. En los días de hemodiálisis, VEMLIDY® se debe administrar tras completar el tratamiento de hemodiálisis. No pueden darse recomendaciones de dosis para pacientes con ClCr < 15 ml/min que no estén recibiendo hemodiálisis. Insuficiencia hepática: No se requiere ajustar la dosis de VEMLIDY® en pacientes con insuficiencia hepática. Población pediátrica: No se ha establecido todavía la seguridad y eficacia de VEMLIDY® en niños menores de 12 años de edad, o que pesen < 35 kg. No se dispone de datos. Estudios clínicos: La eficacia y seguridad de VEMLIDY® en pacientes con hepatitis B crónica se basan en datos de 48 y 96 semanas de dos estudios aleatorizados, con doble ciego, controlados con tratamiento activo, GS-US-320-0108 ("Estudio 108") y GS-US-320-0110 ("Estudio 110"). La seguridad de VEMLIDY® también está respaldada por los datos agrupados de los pacientes de los estudios 108 y 110 que permanecieron con tratamiento ciego desde la semana 96 hasta la semana 120 y adicionalmente de los pacientes de la fase abierta de los estudios 108 y 110 desde la semana 96 hasta la semana 120 (N = 361 permanecieron con VEMLIDY®; N = 80 cambiaron desde tenofovir disoproxilo fumarato a VEMLIDY® en la semana 96). En el Estudio 108, se aleatorizaron pacientes AgHBe negativo con y sin tratamiento previo y con la función hepática compensada, en una proporción de 2:1, para recibir VEMLIDY® (25 mg; N = 285) una vez al día o bien tenofovir disoproxil fumarato (300 mg; N = 140) una vez al día. La media de edad fue de 46 años, el 61 % eran varones, el 72 % eran asiáticos, el 25 % eran de raza blanca y el 2 % (8 pacientes) eran negros; un 24 %, 38 % y 31 % tenían genotipo B, C y D del VHB, respectivamente. Un 21 % había recibido tratamiento previo (tratamiento previo con antivirales orales, incluyendo entecavir (N = 41), lamivudina (N = 42), tenofovir disoproxil fumarato (N = 21) u otros (N = 18)). En el momento basal, la media de ADN del VHB en plasma era de 5,8 log10 UI/ml, la media de ALT sérico era de 94 U/l y un 9 % de los pacientes tenían antecedentes de cirrosis. En el Estudio 110, se aleatorizó a pacientes AgHBe positivo con y sin tratamiento previo y con la función hepática compensada, en una proporción de 2:1, para recibir VEMLIDY® (25 mg; N = 581) una vez al día o bien tenofovir disoproxil fumarato (300 mg; N = 292) una vez al día. La media de edad fue de 38 años, el 64 % eran varones, el 82 % eran asiáticos, el 17 % eran de raza blanca y < 1 % (5 pacientes) eran negros. Un 17 %, 52 % y 23 % tenían genotipo B, C y D del VHB, respectivamente. Un 26 % había recibido tratamiento previo (tratamiento previo con fármacos antivirales orales, incluyendo adefovir (N = 42), entecavir (N = 117), lamivudina (N = 84), telbivudina (N = 25), tenofovir disoproxil fumarato (N = 70) u otros (N = 17)). En el momento basal, la media de ADN del VHB en plasma era de 7,6 log10 UI/ml, la media de ALT sérico era de 120 U/l y un 7 % de los pacientes tenían antecedentes de cirrosis. El parámetro principal de la eficacia en ambos estudios fue la proporción de pacientes con concentraciones plasmáticas de ADN del VHB inferiores a 29 UI/ml en la semana 48. VEMLIDY® cumplió el criterio de no inferioridad al alcanzar un ADN del VHB inferior a 29 UI/ml comparado con tenofovir disoproxil fumarato. Los resultados de tratamiento del Estudio 108 y del Estudio 110 hasta la semana 48 están representados en la Tabla 2 y en la Tabla 3. (Ver Tabla 2 y 3.)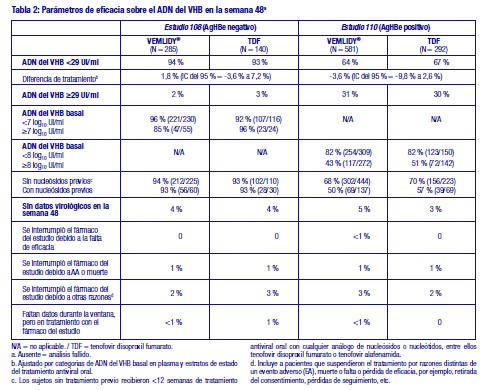
Experiencia más allá de las 48 semanas en el Estudio 108 y en el Estudio 110: En la semana 96, la supresión viral, así como las respuestas bioquímicas y serológicas, se mantuvieron con el tratamiento continuado con tenofovir alafenamida (ver Tabla 4).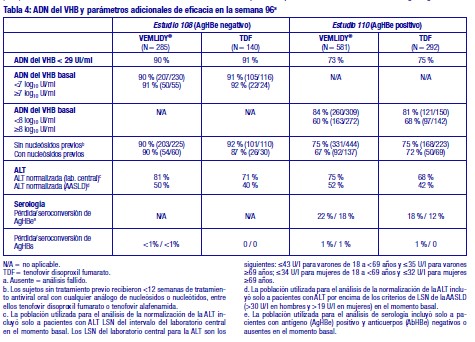
Cambios en las medidas de la densidad mineral ósea: En ambos estudios, tenofovir alafenamida se asoció con menores disminuciones porcentuales en la densidad mineral ósea (DMO; determinada por análisis de absorciometría de rayos x de energía dual [DEXA] de la cadera y de la columna lumbar) más pequeñas que con tenofovir disoproxil fumarato tras 72 semanas de tratamiento. En los pacientes que permanecieron con tratamiento ciego más allá de la semana 96, el cambio porcentual medio en la DMO en cada grupo en la semana 120 fue similar al de la semana 96. En la fase abierta de ambos estudios, el cambio porcentual medio en la DMO desde la semana 96 hasta la semana 120 en los pacientes que permanecieron con VEMLIDY® fue del +0,6 % en la columna lumbar y del 0 % en la cadera total, en comparación con el +1,7 % en la columna lumbar y el +0,6 % en la cadera total en aquellos que cambiaron de tenofovir disoproxilo fumarato a VEMLIDY® en la semana 96. Cambios en las medidas de la función renal: En ambos estudios, tenofovir alafenamida se asoció con cambios en los parámetros de seguridad renal (la mediana de las reducciones más pequeñas en el ClCr estimado mediante Cockcroft-Gault y la mediana de los aumentos porcentuales más pequeños en la proporción de proteína de unión al retinol en orina frente a creatinina y de b-2-microglobulina en orina frente a creatinina) que con tenofovir disoproxil fumarato tras 72 semanas de tratamiento (ver también la sección 7.1). En los pacientes que permanecieron con tratamiento ciego más allá de la semana 96 en los estudios 108 y 110, el cambio con respecto al momento basal en los valores de los parámetros analíticos renales en cada grupo en la semana 120 fueron similares a los de la semana 96. En la fase abierta de los estudios 108 y 110, el cambio medio (±DE) en la creatinina sérica desde la semana 96 hasta la 120 fue de -0,002 (0,10) mg/dl en aquellos que permanecieron con VEMLIDY®, en comparación con -0,008 (0,09) mg/dl en aquellos que cambiaron de tenofovir disoproxilo fumarato a VEMLIDY® en la semana 96. En la fase abierta, la mediana de cambio en la FGe desde la semana 96 hasta la semana 120 fue de -0,6 ml/minuto en los pacientes que permanecieron con VEMLIDY®, en comparación con +1,8 ml/minuto en los pacientes que cambiaron de tenofovir disoproxilo fumarato a VEMLIDY® en la semana 96.
Conservación.
Mantener a temperatura ambiente hasta 30°C. Conservar en su envase original. Proteger de la humedad. Mantener el frasco bien cerrado.
Sobredosificación.
En caso de sobredosis, se debe vigilar al paciente por si hay evidencia de toxicidad (ver Reacciones adversas). El tratamiento de la sobredosis de VEMLIDY® consiste en medidas generales de apoyo, incluyendo la monitorización de las constantes vitales, así como la observación del estado clínico del paciente. Tenofovir se elimina eficazmente mediante hemodiálisis, con un coeficiente de extracción de aproximadamente el 54 %. Se desconoce si tenofovir puede eliminarse mediante diálisis peritoneal.
Presentación.
Envases conteniendo 30 comprimidos recubiertos. El frasco contiene un gel desecante para protegerlo de la humedad.
Revisión.
12/2020. G00200401-00.