ALECENSA®
ROCHE
Agente antineoplásico, inhibidor de la proteína quinasa.
Composición.
Cada cápsula dura contiene 150 mg de alectinib (equivalente a 161,33 mg de alectinib clorhidrato), en un excipiente compuesto por: Masa de relleno de la cápsula: Lactosa monohidratada 33,67 mg, hidroxipropilcelulosa 15,00 mg, laurilsulfato de sodio 75,00 mg, croscarmelosa cálcica (carboximetilcelulosa cálcica) 43,35 mg y estearato de magnesio 1,65 mg. Cubierta de la cápsula: Carragenina 0,25 mg, cloruro de potasio 0,42 mg, dióxido de titanio (E171) 4,20 mg, cera de carnauba < 0,10 mg, almidón de maíz < 0,10 mg, hipromelosa (hidroxipropilmetilcelulosa) 61,63 mg y tinta de impresión. La tinta de impresión está compuesta de (entre corchetes se indican las cantidades relativas): óxido de hierro rojo E172: [5,0%], óxido de hierro amarillo E172: [5,5%], indigotina [carmín de índigo] E132: [13,0%], cera de carnauba [3,5%], goma laca blanca [21,0%], monooleato de glicerilo [0,5%], 1-butanol [30,5%] y alcohol etílico deshidratado [21,0%].
Farmacología.
Código ATC: L01X E36. Grupo farmacoterapéutico: Agente antineoplásico, inhibidores de la proteína quinasa. Propiedades farmacodinámicas: Mecanismo de acción: Alectinib es un inhibidor de la tirosina quinasa dirigido a ALK y RET. En estudios preclínicos, alectinib inhibió la fosforilación de ALK y la activación mediada por ALK de las proteínas de la cascada de señalización hacia abajo STAT3 (transductor de señal y activador de la transcripción 3) y AKT (proteína quinasa 3) y redujo la viabilidad de las células tumorales en varias estirpes celulares que albergan fusiones de ALK, amplificaciones o mutaciones activadas. El metabolito activo principal del Alectinib, M4, demostró tener una potencia y actividad similares in vitro. Se ha comprobado que alectinib y M4 tienen actividad in vitro e in vivo contra varias formas mutantes de la enzima ALK, con inclusión de algunas mutaciones presentes en los tumores CPCNP en pacientes que presentaron progresión con crizotinib. La administración de alectinib produjo actividad antitumoral y prolongó la sobrevida en modelos murinos a los que se les implantó tumores con fusiones de ALK, con inclusión de modelos murinos a los que se les implantó estirpes de células tumorales impulsadas por ALK en el espacio intracraneal. Eficacia clínica y seguridad: Se realizaron dos estudios clínicos multicéntricos de un único grupo de tratamiento para determinar la seguridad y eficacia de Alecensa (Estudios 1 y 2). En ambos estudios, se incluyeron pacientes con CPCNP ALK positivo metastásica a avanzada a nivel local, que habían presentado progresión con crizotinib, con CPCNP con ALK positiva registrado basándose en las pruebas aprobadas por la FDA y con ECOG PS de 0-2. De acuerdo a los criterios de selección, se incorporaron pacientes con tratamientos previos de quimioterapia y radioterapia en el SNC (sistema nervioso central) tras determinar que las metástasis en el SNC se mantuvieron estables por lo menos durante dos semanas. Todos los pacientes recibieron una dosis de 600 mg de Alecensa por vía oral dos veces por día. El criterio de valoración primario de la eficacia en ambos estudios fue la tasa de respuesta objetiva (ORR, objective response rate) de acuerdo con el método RECIST v1.1 (Response Evaluation Criteria in Solid Tumours - Criterios de Evaluación de Respuesta en Tumores Sólidos) según lo evaluado por el Comité de Revisión Independiente (CRI). Otros criterios de valoración según la evaluación del CRI fueron la duración de la respuesta (DOR, duration of response), ORR del SNC y DOR del SNC. El Estudio 1 se llevó a cabo en América del Norte e incluyó un total de 87 pacientes. Las características demográficas y patológicas iniciales en el Estudio 1 fueron: mediana de edad de 54 años (rango de 29 a 79 años, 18% de 65 años o más), 84% caucásicos, 8% asiáticos, 55% mujeres, 35% ECOG PS 0 y 55% ECOG PS I, 100% nunca fumaron o eran ex fumadores, 99% estadio IV, 94% adenocarcinoma y 74% habían recibido quimioterapia previa. Los sitios más comunes de metástasis extratorácica fueron: 60% SNC (65% de los pacientes recibieron radiación al SNC), 43% ganglios linfáticos, 36% hueso y 34% hígado. El Estudio 2 fue internacional e incorporó un total de 138 pacientes. Las características demográficas y patológicas al inicio en el Estudio 2 fueron: mediana de edad de 52 años (rango de 22 a 79 años, el 10% de 65 años de edad o más), 67% caucásicos y 26% asiáticos, 56% mujeres, 32% ECOG PS 0 y 59% ECOG PS 1, 98% no fumadores o ex fumadores, 99% estadio IV, 96% adenocarcinoma y 80% habían recibido quimioterapia previa. Los sitios más comunes de metástasis extratorácica fueron: 61% SNC (73% de los pacientes recibieron radiación al SNC), 51% hueso, 38% ganglios linfáticos y 30% hígado. En la Tabla 1, se presenta un resumen de los resultados de eficacia de los Estudios 1 y 2 para todos los pacientes tratados. La mediana de la duración del seguimiento del Estudio 1 fue de 4,8 meses tanto para las evaluaciones del CRI como del investigador y en el Estudio 2, de 10,9 meses para las del CRI y 7,0 meses para las evaluaciones del investigador. Todas las respuestas son parciales.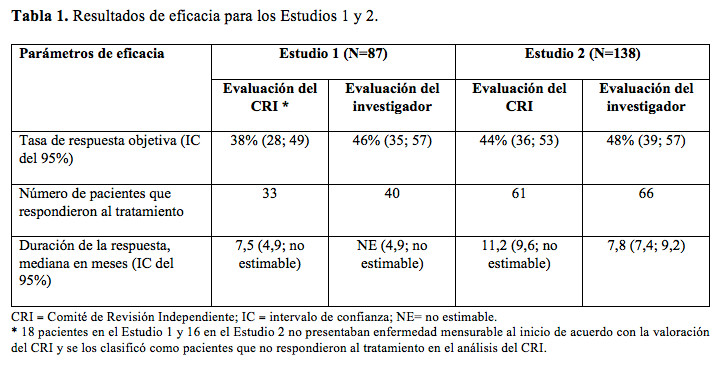
En la Tabla 2, se presenta el resumen de una evaluación de la ORR y de la duración de la respuesta para la metástasis en el SNC en el subgrupo compuesto por 51 pacientes en los Estudios 1 y 2 con lesiones mensurables en el SNC al inicio según RECIST v1.1.; 35 pacientes (69%) con lesiones mensurables en el SNC habían recibido radioterapia cerebral, entre los que se incluyó 25 (49%) que completaron tratamiento radiante por lo menos 6 meses antes de comenzar el tratamiento con Alecensa. Se observó que las respuestas no se vieron afectadas por la radioterapia previa dirigida al cerebro.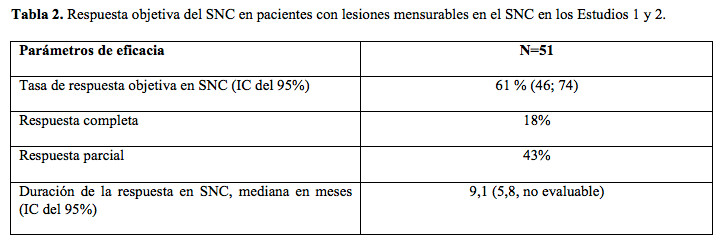
Electrofisiología cardíaca: Se realizó la evaluación de la capacidad de alectinib para expandir el intervalo QT en 221 pacientes a los que se les administró la dosis de 600 mg de Alecensa dos veces por día en estudios clínicos. Alecensa no produjo una prolongación del intervalo QTc (QT corregido para la frecuencia cardíaca) clínicamente relevante. Un paciente obtuvo un valor QTcF (intervalo QTc corregido mediante la fórmula de Fridericia) máximo posterior al valor de referencia superior a los 500 ms y otro paciente presentó un cambio máximo del QTcF desde la toma de los valores de referencia superior a los 60 ms. Población pediátrica: Dado que el fármaco Alecensa ha recibido designación de droga huérfana por los Estados Unidos correspondiente a la presente solicitud, el mismo se encuentra exento de ser evaluado en cuanto a la seguridad y efectividad para todas las solicitudes para nuevos principios activos, nuevas indicaciones, nuevas formas farmacéuticas, nuevos regímenes de dosis o nuevas vías de administración para las indicaciones alegadas en pacientes pediátricos, excepto cuando este requisito no aplicare o fuere pospuesto o se renunciare a él. Propiedades farmacocinéticas: La farmacocinética de alectinib y su metabolito activo principal, M4, ha sido caracterizada en pacientes con CPCNP ALK positivo y en pacientes sanos. En pacientes con CPCNP con ALK positiva, la media geométrica (% del coeficiente de variación) de la concentración máxima (Cmáx, ss) en estado estacionario para alectinib fue de 665 ng/ml (44%) y para M4 de 246 ng/ml (45%) con una tasa de concentración desde el pico hasta el punto mínimo de 1,2. La media geométrica del área bajo la curva en estado estacionario de 0 a 12 horas (ABC 0-12h, ss) para alectinib fue de 7,430 ng·h/ml (46%) y para M4 de 2,810 ng·h/ml (46%). La exposición a alectinib es proporcional a la dosis para todo el rango de dosis de 460 mg a 900 mg (es decir, de 0,75 a 1,5 veces la dosis recomendada aprobada) cuando se lo acompaña con alimentos. Alectinib y M4 alcanzaron las concentraciones en estado estacionario al día 7. La media geométrica de la acumulación fue aproximadamente 6 veces superior para alectinib y M4. Absorción: La concentración máxima de alectinib se alcanzó a las 4 horas de la administración de la dosis de Alecensa de 600 mg dos veces al día cuando se la acompañó con alimentos en pacientes con CPCNP ALK positivo. La biodisponibilidad absoluta de alectinib fue de 37% (IC del 90%: 34%; 40%) cuando se tomó la dosis con alimentos. Las comidas ricas en grasa y calorías incrementaron la exposición combinada (ABC0-inf) de alectinib más M4 en 3,1 veces (IC del 90%: 2,7; 3,6) luego de la administración oral de una única dosis de 600 mg de Alecensa. Distribución: El volumen aparente de distribución es de 4,016 litros para alectinib y de 10,093 litros para M4. Alectinib y M4 se unen a las proteínas plasmáticas humanas en más del 99%, independientemente de la concentración del fármaco. Las concentraciones de alectinib en el líquido cefalorraquídeo en pacientes con CPCNP ALK positivo fueron similares a las concentraciones plasmáticas libres estimadas de alectinib. Los estudios in vitro sugieren que alectinib no es un sustrato de la P-glicoproteína (P-gp), pero que M4 es un sustrato de la P-gp. Alectinib y M4 no son sustratos de la BCPR (breast cáncer resistance protein - proteína de resistencia del cáncer de mama), OATP (organic anion-transporting polypeptide - polipéptido transportador de aniones orgánicos), 1B1 o OATP1B3. Biotransformación (Metabolismo): Alectinib es metabolizado por CYP3A4 a su principal metabolito activo de M4. La media geométrica de la tasa de exposición de metabolito/fármaco original en estado estacionario es de 0,40. Posteriormente, M4 es metabolizado por CYP3A4. Alectinib y M4 son las porciones principales presentes en plasma y representan el 76% de la radioactividad total. Eliminación: La depuración aparente (CL/F) es de 81, 9 l/hora para alectinib y de 217 1/hora para M4. La media geométrica de la semivida de eliminación es de 33 horas para alectinib y de 31 horas para M4 en pacientes con CPCNP con ALK positiva. Excreción: El 98% de la radioactividad se excretó en las heces luego de la administración por vía oral de una única dosis de alectinib marcada con radioisótopos acompañada por alimentos. El 84% de la dosis se excretó en las heces como alectinib inalterado y el 6% de la dosis como M4. La excreción de la radioactividad en la orina no superó el 0,5% de la dosis administrada de alectinib marcada por radioisótopos. Farmacocinética en poblaciones especiales: Poblaciones específicas: La edad, peso corporal, insuficiencia hepática leve, insuficiencia renal de leve a moderada (clearence de creatinina de 30 a 89 ml/min), raza (blanca, asiática, otra) y el sexo no afectaron de manera clínicamente significativa la exposición sistémica de alectinib y M4. No se han realizado estudios sobre la farmacocinética del alectinib en pacientes con insuficiencia renal severa, enfermedad renal terminal o insuficiencia hepática de moderada a grave (véase Dosificación; Poblaciones especiales). Datos preclínicos sobre seguridad: Carcinogenicidad: No se han llevado a cabo estudios para evaluar la carcinogenicidad de alectinib. Mutagenicidad: Alectinib no resultó mutagénico in vitro en la prueba de mutación bacteriana inversa (Ames), pero sí lo fue en una mayor cantidad de micronúcleos en la prueba de micronúcleos de médula ósea en ratas. El mecanismo de inducción de los micronúcleos fue una segregación anormal cromosómica anormal (aneugenicidad) y no un efecto clastogénico sobre los cromosomas. Trastornos de la fertilidad: No se han realizado estudios en animales para evaluar el efecto que alectinib produce en la fertilidad. Teratogenicidad: No se observaron efectos adversos en los órganos reproductores de hombres y mujeres en los estudios de toxicología general realizados en ratas y monos.
Indicaciones.
Alecensa está indicado para el tratamiento de pacientes con cáncer de pulmón de células no pequeñas (CPCNP) metastásico con quinasa de linfoma anaplásico (ALK) positiva que presentaron progresión o intolerancia al tratamiento con crizotinib. La presente indicación se basa en la tasa de respuesta tumoral y en la duración de la respuesta (véase Farmacología - Propiedades farmacodinámicas). La continuidad de la aprobación de esta indicación queda sujeta a la verificación y descripción de los beneficios clínicos en los estudios confirmatorios.
Dosificación.
La dosis recomendada de Alecensa es de 600 mg por vía oral dos veces por día con alimentos (véase Farmacología). Administrar Alecensa hasta que se produzca la progresión de la enfermedad o hasta que el nivel de toxicidad sea inadmisible. No abra ni disuelva el contenido de la cápsula. Si omite una dosis de Alecensa o si vomita después de tomar una dosis de Alecensa, debe tomar la siguiente dosis en la hora pautada. Modificación de la dosis por reacciones adversas: En la Tabla 3, se puede observar el programa de reducción de la dosis de Alecensa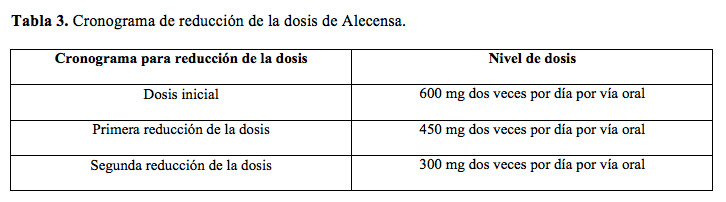
Se deberá discontinuar el tratamiento si el paciente no tolera la dosis de 300 mg dos veces por día. En la Tabla 4, se presentan las recomendaciones para la modificación de la dosis de Alecensa por reacciones adversas.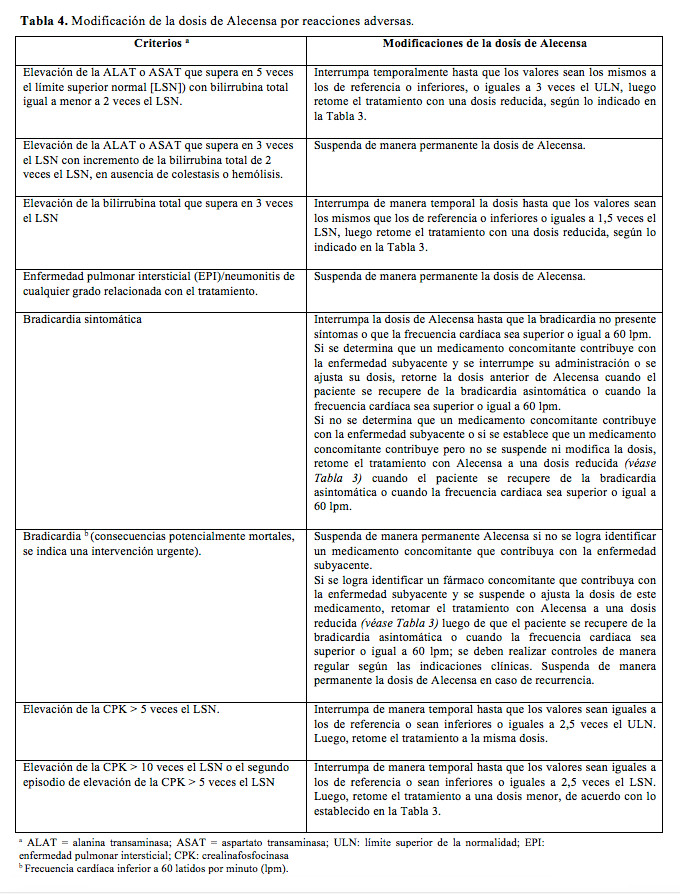
Poblaciones especiales: Población pediátrica: No se han determinado la seguridad y efectividad de Alecensa en pacientes pediátricos. Datos en animales: Aún no se han realizado estudios de alectinib en animales jóvenes. En los estudios de toxicología general, el tratamiento con alectinib en ratas que dio como resultado exposiciones iguales o superiores a aproximadamente 4,5 veces las exposiciones en seres humanos tratados con 600 mg de alectinib dos veces por día provocó cambios en los dientes y huesos en crecimiento. Se produjo decoloración y alteración en el tamaño de los dientes, así como el desajuste histopatológico de los ameloblastos y de las capas odontoblásticas. Asimismo, se advirtió una disminución en el hueso trabecular y un aumento de la actividad de los osteoclastos en el fémur y el esternón. Pacientes de edad avanzada: Los estudios clínicos sobre Alecensa no contaron con la cantidad suficiente de pacientes de 65 años de edad o más como para determinar si estos responden al tratamiento igual que los más jóvenes. Pacientes con insuficiencia renal: No se recomienda un ajuste de la dosis en pacientes con insuficiencia renal de leve a moderada. No se han efectuado estudios sobre la seguridad de Alecensa en pacientes con insuficiencia renal grave (depuración de la creatinina inferior a 30 ml/min) o con enfermedad renal terminal (véase Farmacología). Pacientes con insuficiencia hepática: No se recomienda un ajuste de la dosis en pacientes con insuficiencia hepática leve (bilirrubina total inferior o igual al ULN (upper limit of normal - límite superior de la normalidad) y aspartato aminotransferasa (AST) superior al ULN o bilirrubina total superior a 1,0-1,5 veces el ULN y cualquier AST). No se han realizado estudios sobre la seguridad de Alecensa en pacientes con insuficiencia hepática moderada o grave (véase Farmacología). Formas de administración: Alecensa se administra por vía oral. Las cápsulas duras deben ser ingeridas enteras con las comidas y no deben abrirse ni disolverse.
Contraindicaciones.
Hipersensibilidad a cualquiera de los componentes de la formulación.
Reacciones adversas.
Las siguientes reacciones adversas se explican más detalladamente en otras secciones del prospecto: Hepatotoxicidad (véase Precauciones). Enfermedad pulmonar intersticial (EPI)/neumonitis (véase Precauciones). Bradicardia (véase Precauciones). Mialgia severa y elevación de la creatina-fosfocinasa (CPK) (véase Precauciones). Toxicidad embriofetal (véase Precauciones). Experiencia en estudios clínicos: Dado que los estudios clínicos se llevan a cabo en una gran cantidad de condiciones variables, no es posible comparar de manera directa las tasas de reacciones adversas de un determinado fármaco con las de otros medicamentos y puede que estas no coincidan con las tasas que se observan en la práctica. La seguridad de Alecensa se evaluó en 253 pacientes con cáncer de pulmón de células no pequeñas (CPCNP) con quinasa de linfoma anaplásico (ALK) positiva que recibieron 600 mg de Alecensa por vía oral dos veces por día en dos estudios clínicos, el Estudio l y el Estudio 2. La mediana de duración de la exposición a Alecensa fue de 9,3 meses. Se expuso a 169 pacientes (67%) a Alecensa por más de 6 meses y a 100 (40%) por más de un año. Las características de la población fueron: mediana de edad de 53 años, edad inferior a 65 años (86%), mujeres (55%), caucásicos (74%), asiáticos (18%), con histología de adenocarcinoma CPCNP (96 %), nunca fumó o ex fumador (98%), ECOG PS (Eastern Cooperative Oncological Group Performance Status - estado de desempeño del paciente del Eastern Cooperative Oncological Group) 0 o 1 (91%) y quimioterapia previa (78%). Ocurrieron reacciones adversas serias en el 19% de los pacientes. Las reacciones adversas serias informadas más frecuentes fueron: embolia pulmonar (1,2%), disnea (1,2%) e hiperbilirrubinemia (1,2%). Se presentaron reacciones adversas fatales en el 2,8% de los pacientes, entre las cuales se encontraban: hemorragia (0,8%), perforación intestinal (0,4%), disnea (0,4%), embolia pulmonar (0,4%) y endocarditis (0,4%). Se suspendió Alecensa de manera permanente en el 6% de los pacientes debido a reacciones adversas. Las reacciones adversas más frecuentes que llevaron a la suspensión permanente de Alecensa fueron hiperbilirrubinemia (1,6%), e incremento en los niveles de ALAT (1,6%) y aumento en los niveles de ASAT (1,2%). En total, el 23% de los pacientes que comenzó el tratamiento en la dosis recomendada necesitó al menos una reducción de la dosis. La mediana de tiempo hasta la primera disminución de la dosis fue de 48 días. Las reacciones adversas más frecuentes que causaron la reducción o interrupción de la dosis fueron las elevaciones de la bilirrubina (6%), CPK (4,3%), ALAT (4,0%) y ASAT (2,8%) y vómitos (2,8%). En la Tabla 5, encontrará un resumen de las reacciones adversas de los Estudios 1 y 2.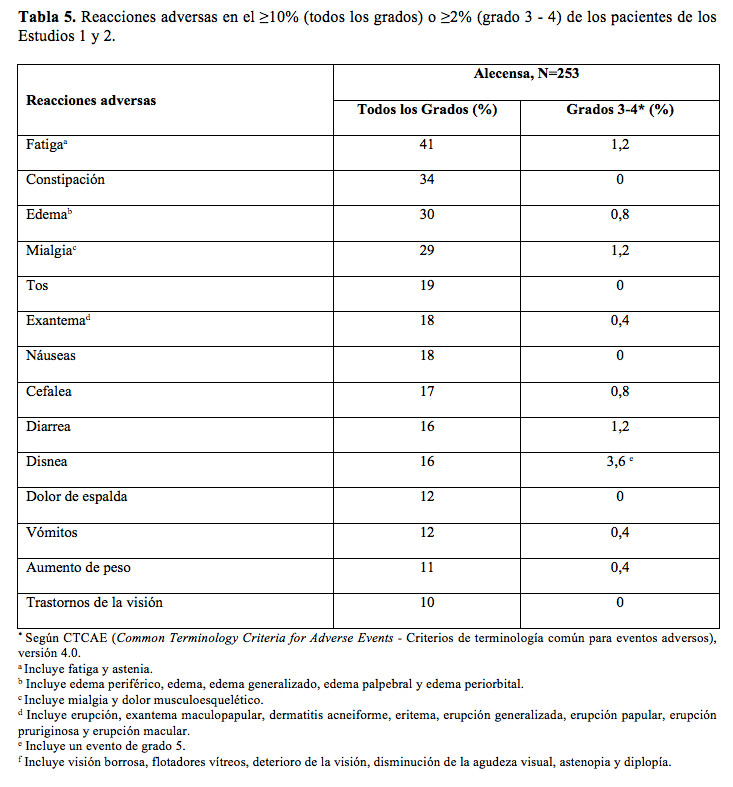
Información adicional sobre seguridad de la experiencia en estudios clínicos: El 9,9% de los pacientes tratados con Alecensa experimentó fotosensibilidad en las Estudios 1 y 2. Se les recomendó no exponerse al sol y usar protector solar de amplio espectro. La incidencia de fotosensibilidad de Grado 2 fue de 0,4%. El resto de los eventos fue de Grado 1. En la Tabla 6 se presenta un resumen de las anomalías de laboratorio para Alecensa en los Estudios 1 y 2.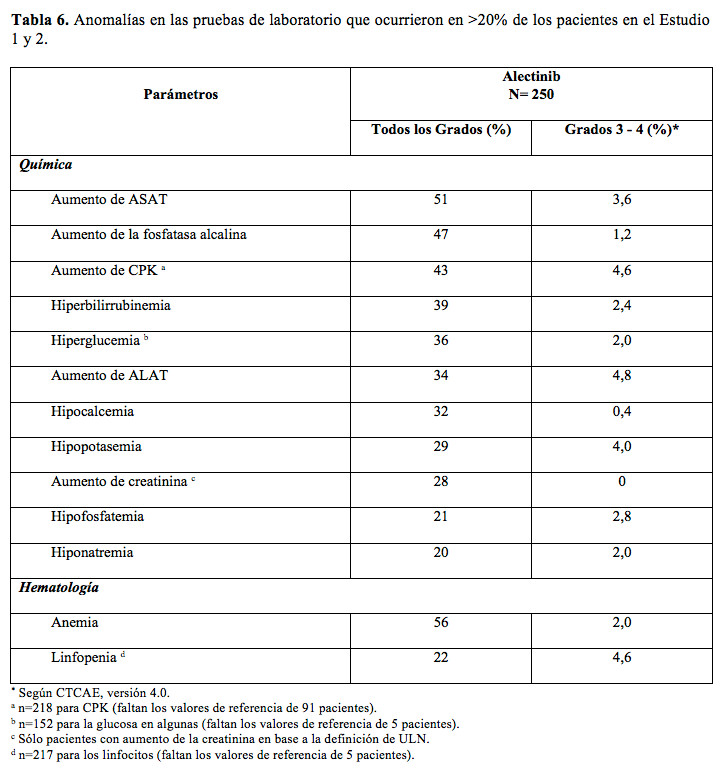
Comunicación de reportes de reacciones adversas: Es importante comunicar las presuntas reacciones adversas después de la autorización del medicamento. Esto permite la monitorización continua de la relación riesgo/beneficio. Se solicita a los profesionales de la salud informar sobre cualquier sospecha de eventos adversos asociados con el uso de Alecensa® al Área de Farmacovigilancia de Roche al siguiente teléfono 0800-77-ROCHE (76243). En forma alternativa, esta información puede ser reportada ante ANMAT. Ante cualquier inconveniente con el producto, el paciente puede llenar la ficha que está en la Página Web de la ANMAT: http://www.anmat.gov.ar/farmacovigilancia/Notificar.asp o llamar a ANMAT responde al 0800-333-1234.
Precauciones.
Enfermedad pulmonar intersticial (EPI)/Neumonitis: Uno (0,4%) de 253 pacientes expuestos a Alecensa en los estudios clínicos presentó EPI grave (Grado 3). Se deberá analizar lo antes posible a aquellos pacientes con empeoramiento de los síntomas respiratorios que indique un diagnóstico de EPI/neumonía (por ejemplo, disnea, tos o fiebre). Los pacientes deberán ser informados sobre los riesgos de EPI/neumonitis grave. Se deberá aconsejar que se comuniquen inmediatamente con un profesional de la salud para informar si se presentan nuevos síntomas de problemas respiratorios o si estos síntomas empeoran. Interrumpa el tratamiento con Alecensa inmediatamente si el paciente ha sido diagnosticado con EPI/neumonitis y suspéndalo de manera permanente si no se han determinado otras causas posibles para tal diagnóstico (véanse Dosificación y Reacciones Adversas). Hepatotoxicidad: Las elevaciones de la AST que superen en más de 5 veces el ULN (upper limit of normal - límite superior de la normalidad) ocurrieron en 3,6% de los pacientes y las elevaciones de la ALT que superan en más de 5 veces el ULN se manifestaron en el 4,8%. Las elevaciones de la bilirrubina que superan en más de 3 veces el ULN ocurrieron en 2,8% de los pacientes. La mayoría de estos eventos (el 73% con elevación de las transaminasas hepáticas y el 49% con elevaciones de la bilirrubina) se produjeron durante los primeros 2 meses de tratamiento. Cuatro pacientes interrumpieron la dosis de Alecensa debido a elevaciones de la ASAT y/o ALAT de Grados 3-4 y tres suspendieron el tratamiento con Alecensa debido a elevaciones de la bilirrubina de Grado 3. Se documentó mediante una biopsia del hígado que 2 pacientes con elevaciones de ASAT/ALAT de Grados 3-4 habían sufrido daño hepático inducido por medicamentos. Los pacientes deberán ser informados sobre los signos y síntomas de las elevaciones de la bilirrubina y de la transaminasa hepática. Se deberá aconsejar que se comuniquen con un profesional de la salud inmediatamente si presentan signos o síntomas de elevaciones de la bilirrubina o de la transaminasa hepática. Se deberá monitorear las pruebas de función hepática, con inclusión de ALAT, ASAT y bilirrubina cada dos semanas durante los primeros dos meses y luego de manera regular durante el transcurso del tratamiento. Los pacientes con elevaciones de transaminasa o de bilirrubina deberán realizarse evaluaciones con mayor frecuencia. En virtud de la gravedad de las reacciones adversas al fármaco, interrumpa el tratamiento con Alecensa y retómelo con una dosis menor o suspéndalo de manera permanente, según se indica en la Tabla 4 (véase Dosificación). Mialgia severa y elevación de la creatina fosfoquinasa (CPK): El 29% de los pacientes del Estudio 1 y del Estudio 2 presentaron mialgia o dolor musculoesquelético. La incidencia de mialgia/dolor musculoesquelético de Grado 3 fue del 1,2%. El 0,8% de los pacientes necesitó una modificación de la dosis a causa de la mialgia/dolor musculoesquelético. El 43% de 218 pacientes en los Estudios l y 2 manifestaron elevaciones de la CPK con datos de CPK de laboratorio disponibles. La incidencia de las elevaciones de CPK de Grado 3 fue del 4,6%. La mediana de tiempo hasta la elevación de CPK de Grado 3 fue de 14 días (rango intercuartil de 13-14 días). Se realizó la modificación de la dosis en el 0,5% de los pacientes como consecuencia de la elevación de la CPK. Se deberá informar a los pacientes sobre los signos y síntomas de mialgia, entre las que se incluye dolor, sensibilidad o debilidad muscular sin razón aparente y/o persistente. Se debe aconsejar a los pacientes que comuniquen cualquier dolor, sensibilidad o debilidad musculares sin razón aparente. Evalúe los niveles de CPK cada dos semanas durante el primer mes de tratamiento y según indicaciones clínicas en pacientes que han informado síntomas. En virtud de la gravedad de la elevación de CPK, interrumpa el tratamiento con Alecensa y luego retómelo o reduzca la dosis (véase Dosificación). Bradicardia: El tratamiento con Alecensa puede provocar bradicardia sintomática. Se han informado casos de bradicardia (7,5%) en pacientes tratados con Alecensa. El 20% de 221 pacientes tratados con Alecensa en los que se realizaron electrocardiogramas seriados obtuvo frecuencias cardíacas inferiores a los 50 latidos por minuto (lpm). Controlar la frecuencia cardíaca y la presión arterial de manera regular. No es necesario realizar una modificación de la dosis cuando se presenta bradicardia asintomática. Los pacientes deberán ser informados sobre los síntomas de bradicardia, como mareos o la sensación de mareos y síncope, que pueden aparecer durante la administración de Alecensa. Se deberá aconsejar que se comuniquen con un profesional de la salud para informar si se presentan estos síntomas y sobre el uso de fármacos para el corazón o la tensión arterial. En el caso de un diagnóstico de bradicardia sintomática que no resulta ser potencialmente fatal, interrumpa la dosis de Alecensa hasta que los síntomas desaparezcan o hasta que la frecuencia cardíaca sea igual o superior a 60 lpm y evite los medicamentos concomitantes que se sabe producen bradicardia, así como los agentes antihipertensivos. Si la enfermedad subyacente se debe a un medicamento concomitante, retome el tratamiento con Alecensa en una dosis reducida (véase Tabla 3) luego de que el paciente se haya recuperado de la bradicardia asintomática o cuando la frecuencia cardíaca sea igual o superior a 60 lpm y realice controles de manera regular según indicaciones clínicas. Si el paciente sufre una recurrencia, suspenda de manera definitiva el tratamiento con Alecensa. Interrumpa permanentemente el tratamiento con Alecensa si se manifiestan casos de bradicardia potencialmente fatales y si no se ha podido determinar que un medicamento concomitante es la causa de este diagnóstico (véase Dosificación). Fotosensibilidad: Se debe asesorar a los pacientes sobre los signos y síntomas de fotosensibilidad. Se deberá aconsejarles que eviten la exposición solar prolongada mientras se encuentren en tratamiento con Alecensa y, por lo menos, durante 7 días posteriores a la interrupción del tratamiento con el fármaco del estudio y recomendar el uso de protector solar. Se deberá indicar el uso de protector solar de amplio espectro contra los rayos ultravioleta A (UVA)/ultravioletas B (UVB) y bálsamo labial (FPS ≥50) para protegerse de posibles quemaduras solares (véase Reacciones adversas). Toxicidad embrionaria-fetal: En virtud de los resultados obtenidos de estudios de animales y de su mecanismo de acción, Alecensa puede ocasionar daño fetal cuando se lo administra a una mujer embarazada. La administración de alectinib en ratas y conejos preñados durante el período de organogénesis produjo toxicidad embriofetal y aborto a dosis tóxicas para la madre, con exposiciones aproximadamente 2,7 veces superiores a las observadas en seres humanos con la dosis de 600 mg de alectinib dos veces por día. Se deberá advertir a las mujeres embarazadas sobre el posible riesgo para el feto. Se debe asesorar a las mujeres en edad fértil sobre la utilización de métodos anticonceptivos durante el tratamiento con Alecensa y durante una semana luego de la toma de la última dosis (véase Precauciones y Farmacología). Se deberá recomendarles que informen a un profesional de la salud sobre cualquier embarazo confirmado o sospecha de embarazo. Se deberá aconsejar a los pacientes masculinos con parejas de sexo femenino en edad fértil sobre el uso de métodos anticonceptivos durante el tratamiento con Alecensa y durante los 3 meses posteriores a la última dosis. Intolerancia a la lactosa: Este medicamento contiene lactosa. Los pacientes con problemas hereditarios raros de intolerancia a la galactosa, una deficiencia congénita de lactosa o una absorción deficiente de glucosa-galactosa no deben tomar este medicamento. Fertilidad, embarazo y lactancia: Anticonceptivos: Mujeres: El tratamiento con Alecensa en mujeres embarazadas puede provocar daño fetal. Se aconseja el uso de métodos anticonceptivos durante el tratamiento y durante la semana posterior a la última dosis de Alecensa para aquellas mujeres en etapa fértil (véase Precauciones). Hombres: En virtud de los hallazgos de genotoxicidad, se recomienda que aquellos hombres cuya pareja se encuentre en edad fértil utilicen métodos anticonceptivos durante el tratamiento con Alecensa y durante los 3 meses posteriores a la toma de la última dosis (véase Farmacología - Datos preclínicos sobre seguridad). Embarazo: De acuerdo con los estudios realizados en animales y su mecanismo de acción, el tratamiento con Alecensa en mujeres embarazadas puede provocar daño fetal (véase Farmacología). No hay información disponible sobre el uso de Alecensa en mujeres embarazadas. La administración de alectinib a ratas y conejos preñados mediante sonda oral durante el período de organogénesis produjo toxicidad embriofetal y aborto a las dosis tóxicas para las madres con exposiciones aproximadamente 2,7 veces superiores a las observadas en seres humanos tratados con la dosis de 600 mg de alectinib dos veces por día (véase Datos en animales). Se debe advertir a las mujeres embarazadas sobre el posible riesgo para el feto. En la población general de los EE. UU., el riesgo estimado por antecedentes de defectos de nacimiento y aborto en embarazos reconocidos clínicamente es de 24% y de 15-20%, respectivamente. Datos en animales: En un estudio preliminar embriofetal realizado en conejos, la administración de alectinib por sonda oral durante el período de organogénesis produjo el aborto o muerte embriofetal total a dosis de 27 mg/kg/día tóxicas para la madre (aproximadamente 2,9 veces el área bajo la curva (ABC0-24h, 55) en seres humanos tratados con la dosis de alectinib de 600 mg dos veces al día) en tres de los seis conejos preñados. Los tres conejos restantes en este grupo parieron unos pocos fetos vivos, los fetos o la placenta presentaron un peso más bajo y padecieron el síndrome de la arteria subclavia de trayecto retroesofágico. En un estudio preliminar de desarrollo embriofetal realizado en ratas, la administración de alectinib durante el período de organogénesis ocasionó la pérdida de la camada en todas las ratas preñadas a dosis de 27 mg/kg/día (aproximadamente 4,5 veces el ABC0-24h, ss estimado en seres humanos tratados con la dosis de alectinib de 600 mg dos veces por día). Las dosis iguales o superiores a 9 mg/kg/día (aproximadamente 2,7 veces el ABC0-24h, ss estimado en seres humanos tratados con la dosis de alectinib de 600 mg dos veces por día) produjo toxicidad para la madre, así como toxicidad en el desarrollo del feto, incluso disminución del peso fetal, dilatación del uréter, cordón tímico, ventrículo pequeño y paredes delgadas del ventrículo, y disminución en el número de vértebras sacras y coccígeas. Lactancia: No hay información disponible sobre la presencia de alectinib o sus metabolitos en la leche humana, los efectos que alectinib puede producir en el lactante o en la producción de leche. Dado que alectinib puede provocar reacciones adversas serias en lactantes, se aconseja no amamantar durante el tratamiento con Alecensa y por una semana luego de la última dosis.
Interacciones.
Efectos de alectinib sobre otros medicamentos: No se esperan efectos clínicamente relevantes en la exposición de midazolam (sustrato sensible de CYP3A) o repaglinida (sustrato sensible de CYP2C8) luego de la administración conjunta con Alecensa. Los estudios in vitro sugieren que alectinib y M4 no producen la inhibición de CYP1A2, 2B6, 2C9, 2C19 o 2D6, pero sí inhiben P-gp y BCRP. Alectinib no causó la inhibición del transporte in vitro de OATP1B1, OATP1B3, OAT1, OAT3 u OCT2. Efectos de otros medicamentos sobre alectinib: No se han comprobado efectos clínicamente relevantes en la exposición de la combinación de alectinib más M4 en los estudios clínicos luego de la administración conjunta de Alecensa con un inhibidor potente de CYP3A (posaconazol), un inductor potente de CYP3A (rifampina) o un fármaco para la reducción de la acidez (esomeprazol).
Conservación.
Las cápsulas duras deben conservarse a temperatura inferior a 30° C. Almacenar en el envase original para protegerlo de la luz y la humedad. Precauciones especiales de eliminación y otras manipulaciones: Este medicamento no debe ser utilizado después de la fecha de vencimiento indicada en el envase. Este medicamento debe ser usado exclusivamente bajo prescripción y vigilancia médica y no puede repetirse sin nueva receta médica. Mantenga los medicamentos fuera del alcance de los niños.
Sobredosificación.
No hay datos disponibles sobre sobredosis. No hay un antídoto específico para tratar la sobredosis con Alecensa. Alectinib y su metabolito activo principal M4 se unen en un 99 % a las proteínas plasmáticas. Por lo tanto, es probable que una hemodiálisis resulte inútil para tratar una sobredosis. Ante la eventualidad de una sobredosificación, concurrir al Hospital más cercano o comunicarse con los Centros de Toxicología: Hospital de Pediatría Dr. Ricardo Gutiérrez: 4962-6666/2247; Policlínico Dr. G. A. Posadas: 4654-6648, 4658-7777; Hospital General de Niños Dr. Pedro de Elizalde: 4300-2115; 4363-2100/2200, Interno 6217.
Presentación.
Naturaleza y contenido del envase: Blíster de aluminio laminado en frío sellado con una lámina de aluminio (ALU-ALU) que contiene 8 cápsulas. Envase con 224 cápsulas duras. Blísteres con 8 cápsulas duras de 150 mg: envase con 224.
Revisión.
Marzo 2017. Aprobación: 28/08/2017. Disp. ANMAT No. DI-2017-9585-APN-ANMAT. (NP+FDA+ANMAT C004/13 y 1° rcp+Shpe+O(T2016-0707)+2° rcp.