ESCLEROFERON®
BIOSIDUS S.A.U
Inmunomodulador.
Composición.
Cada jeringa prellenada de 0,5 ml contiene 30 microgramos (6 millones de UI) de interferón beta 1a. La concentración de interferón beta 1a es de 30 microgramos por 0,5 ml. Composición por unidad de dosis: Interferón beta 1a 30 mg, Arginina clorhidrato 15,8 mg, Polisorbato 20 0,025 mg, Ácido Acético Glacial 0,6 mg, Hidróxido de sodio c.s.p pH: 4,8, Agua para inyectables c.s.p. 0,5 mL. Forma farmacéutica: Solución inyectable. El color de la solución inyectable debe ser límpido e incoloro.
Farmacología.
Propiedades farmacodinámicas: Grupo farmacoterapéutico: interferones, código ATC: L03 AB07. Los interferones son una familia de proteínas naturales producidas por células eucarióticas en respuesta a infecciones víricas y a diferentes inductores de origen biológico. Los interferones son citocinas que median actividades antivíricas, antiproliferativas e inmunomoduladoras. Se han identificado tres clases principales de interferones: alfa, beta y gamma. Los interferones alfa y beta son interferones de tipo I, y el interferón gamma es un interferón de tipo II. Dichos interferones poseen actividades biológicas superpuestas pero claramente distinguibles. También pueden diferir con respecto a su lugar celular de síntesis. El interferón beta se produce en varios tipos de células, incluidos los fibroblastos y macrófagos. El interferón beta natural y Escleroferon® (interferón beta 1a) están glicosilados y tienen una única unión N-grupo carbohidrato complejo. Se sabe que la glicosilación de otras proteínas afecta a su estabilidad, actividad, biodistribución y semivida en la sangre. Sin embargo, no se han definido por completo los efectos del interferón beta que dependen de la glicosilación. Mecanismo de acción: Escleroferon® ejerce sus efectos biológicos mediante su unión a receptores específicos en la superficie de las células humanas. Dicha unión inicia una compleja secuencia de fenómenos intracelulares que conduce a la expresión de numerosos productos y marcadores inducidos genéticamente por el interferón. Entre ellos están la clase I del complejo mayor de histocompatibilidad (CPH), la proteína Mx, la 2' / 5'-oligoadenilato sintetasa, la b2-microglobulina y la neopterina. Algunos de estos productos se han medido en el suero y en la fracción celular de la sangre de pacientes tratados con interferón beta 1a. Después de una sola dosis intramuscular de interferón beta 1a, los niveles séricos de dichos productos permanecen altos durante al menos cuatro días e incluso hasta una semana. Se desconoce si el mecanismo de acción del interferón beta en la EM sigue la misma ruta que los efectos biológicos descritos anteriormente, porque no está bien establecida la fisiopatología de la EM. Eficacia clínica y seguridad: Los efectos del tratamiento con interferón beta 1a intramuscular (IFN beta 1a IM) se demostraron en un único ensayo controlado con placebo en 301 pacientes (IFN beta 1a IM, n = 158; placebo, n = 143) con EM recidivante caracterizada por al menos 2 exacerbaciones en los últimos tres años o al menos una exacerbación en el año anterior a la entrada del paciente en el ensayo, cuando la duración de la enfermedad era inferior a 3 años. En el ensayo clínico se incluyeron pacientes con un EDSS entre 1,0 y 3,5. Debido al diseño del estudio, el seguimiento de los pacientes se realizó en períodos de tiempo variables. 150 pacientes tratados con IFN beta 1a IM completaron un año en estudio y 85 completaron dos años en estudio. En el estudio, el porcentaje acumulado de pacientes que desarrollaron progresión de la incapacidad (según el análisis de la tabla de vida de Kaplan-Meier) al final del período de dos años fue del 35 % en los pacientes tratados con placebo y del 22 % en los pacientes tratados con IFN beta 1a IM. La progresión de la incapacidad se midió como un incremento en la escala ampliada del estado de discapacidad (EDSS, Expanded Disability Status Scale) de 1,0 punto, sostenido durante al menos seis meses. También se demostró una reducción de un tercio del índice anual de recaída. Este último efecto clínico fue observado después de más de un año de tratamiento. Tras un estudio doble ciego aleatorizado y de comparación de dosis con 802 pacientes con EM recidivante (IFN beta 1a IM 30 mg n = 402, IFN beta 1a IM 60 mg n = 400) no se observaron diferencias estadísticamente significativas o tendencias entre las dosis de IFN beta 1a IM de 30 mg y de 60 mg en cuanto a los parámetros clínicos y mediante imagen por resonancia magnética. Los efectos de IFN beta 1a IM en el tratamiento de la EM también se demostraron en un ensayo doble ciego aleatorizado con 383 pacientes (IFN beta 1a IM n = 193, placebo n = 190) con un único acontecimiento desmielinizante asociado con al menos dos lesiones cerebrales compatibles en la resonancia magnética. En el grupo de tratamiento con IFN beta 1a IM se observó una reducción del riesgo de experimentar un segundo acontecimiento. También se observó un efecto sobre los parámetros medidos por resonancia magnética. El riesgo estimado de un segundo acontecimiento fue del 50% en tres años y del 39% en dos años en el grupo de placebo y del 35% (tres años) y 21% (dos años) en el grupo de IFN beta 1a IM. En un análisis post-hoc, aquellos pacientes con una resonancia magnética basal con al menos una lesión realzada con gadolinio y nueve lesiones en T2 tuvieron un riesgo de sufrir un segundo acontecimiento del 56 % en el grupo de placebo y del 21 % en el grupo tratado con IFN beta 1a IM a los dos años. Sin embargo, el impacto del tratamiento precoz con IFN beta 1a IM se desconoce incluso en este subgrupo de alto riesgo, ya que el estudio fue principalmente diseñado para evaluar el tiempo hasta el segundo acontecimiento, en lugar de la evolución a largo plazo de la enfermedad. Además, por ahora no hay una definición bien establecida de pacientes de alto riesgo, aunque un planteamiento más conservador es aceptar al menos nueve lesiones hiperintensas en T2 en el estudio inicial y al menos una nueva en T2 o una nueva lesión realzada con gadolinio sobre una resonancia de seguimiento recogida al menos tres meses después de la inicial. En cualquier caso el tratamiento debe considerarse solamente para pacientes clasificados como de alto riesgo. Población pediátrica: Los datos limitados sobre la eficacia/seguridad de IFN beta 1a IM 15 microgramos por vía intramuscular una vez a la semana (n=8) en comparación con ningún tratamiento (n=8) con un seguimiento durante 4 años mostraron resultados acordes con los observados en adultos, aunque las puntuaciones de EDSS aumentaron en el grupo tratado durante el seguimiento de 4 años; por tanto, esto indica la progresión de la enfermedad. No hay comparación directa con la dosis actualmente recomendada en adultos. Propiedades farmacocinéticas: El perfil farmacocinético de Escleroferon® se ha investigado indirectamente mediante un análisis que mide la actividad antivírica del interferón. Este análisis es limitado, pues es sensible para el interferón pero carece de especificidad para el interferón beta. Las técnicas de análisis alternativas no son lo suficientemente sensibles. Tras la administración intramuscular de IFN beta 1a, el pico de los niveles de actividad antivírica sérica ocurre entre 5 y 15 horas después de la administración y disminuye con una vida media de aproximadamente 10 horas. Con un ajuste adecuado de la tasa de absorción desde el punto de inyección, la biodisponibilidad calculada es de aproximadamente el 40 %. La biodisponibilidad calculada es mayor sin dichos ajustes. La administración subcutánea no puede suplir la administración intramuscular. Datos preclínicos sobre seguridad: Carcinogénesis: no se dispone de datos de carcinogenicidad del interferón beta 1a en animales o humanos. Toxicidad crónica: no se observó respuesta inmune al interferón beta 1a ni signos de toxicidad en un estudio de toxicidad con dosis repetidas de 26 semanas de duración, administrado en monos rhesus por vía intramuscular una vez a la semana, en combinación con otro agente inmunomodulador, un anticuerpo monoclonal ligando CD40. Tolerancia local: no se ha evaluado la irritación intramuscular en animales tras la administración repetida en el mismo punto de inyección. Mutagénesis: se han llevado a cabo pruebas de mutagénesis limitadas pero relevantes. Los resultados han sido negativos. Alteración de la fertilidad: Se han llevado a cabo estudios de fertilidad y desarrollo en el mono rhesus con una forma relacionada del interferón beta 1a. A dosis muy altas, se observaron efectos anovulatorios y abortivos en animales de ensayo. También se han observado efectos reproductivos similares relacionados con la dosis con otras formas de interferones alfa y beta. No se han observado efectos teratogénicos ni efectos en el desarrollo fetal, pero la información disponible sobre los efectos del interferón beta 1a en los períodos perinatal y posnatal es limitada. No hay información disponible sobre los efectos del interferón beta 1a sobre la fertilidad masculina.
Indicaciones.
Escleroferon® está indicado para el tratamiento de Pacientes diagnosticados con esclerosis múltiple (EM) recidivante. En ensayos clínicos, se caracterizó por dos o más exacerbaciones agudas (recaídas) en los últimos tres años sin evidencia de progresión continua entre recaídas; el interferón beta 1a retrasa la progresión de la incapacidad y disminuye la frecuencia de las recaídas. Pacientes con un único acontecimiento desmielinizante con un proceso inflamatorio activo, si es lo bastante grave como para justificar el tratamiento con corticosteroides intravenosos, si se han excluido diagnósticos alternativos y si resultan tener un riesgo elevado para el desarrollo de esclerosis múltiple definida clínicamente (ver sección Farmacología). Se deberá interrumpir el tratamiento con Escleroferon® en los pacientes que desarrollen esclerosis múltiple progresiva.
Dosificación.
El tratamiento debe iniciarse bajo la supervisión de un médico experto en el tratamiento de la enfermedad. Posología: Adultos: la dosis recomendada para el tratamiento de la EM recidivante es de 30 microgramos (0,5 ml de solución) administrados en inyección intramuscular (IM) una vez por semana. No se ha observado ningún beneficio adicional mediante la administración de una dosis más alta (60 microgramos) una vez a la semana. Ajuste de la dosis: A fin de ayudar a los pacientes a reducir la incidencia y gravedad de los síntomas pseudogripales (ver Reacciones adversas), al iniciarse el tratamiento puede efectuarse una fase de ajuste de la dosis. Escleroferon® puede iniciarse en incrementos de ¼ de dosis por semana hasta alcanzar la dosis completa (30 microgramos/semana) a la cuarta semana. Una pauta de ajuste de la dosis alternativa consiste en iniciar la terapia con aproximadamente ½ dosis, una vez por semana, antes de pasar a la dosis total. Para obtener una eficacia adecuada se debe alcanzar y mantener una dosis de 30 mg una vez por semana tras completarse el período inicial de ajuste de la dosis. Antes de la inyección, y durante un período adicional de 24 horas tras cada inyección, se aconseja el uso de un analgésico antipirético para disminuir los síntomas pseudogripales asociados con la administración de Escleroferon®. Estos síntomas generalmente se presentan durante los primeros meses de tratamiento. Población pediátrica: Todavía no se ha establecido la seguridad y eficacia del interferón beta 1a en adolescentes de 12 a 16 años. Los datos actualmente disponibles están descritos en Reacciones adversas y Farmacología, sin embargo no se puede hacer una recomendación posológica. No se ha establecido todavía la seguridad y eficacia del interferón beta 1a en niños menores de 12 años. No se dispone de datos. Pacientes de edad avanzada: Los ensayos clínicos no han incluido un número de pacientes de 65 años o más suficiente para determinar si responden de forma distinta que los más jóvenes. Sin embargo, teniendo en cuenta el modo de eliminación del principio activo, no hay motivos teóricos para ajustar la dosis en pacientes de edad avanzada. Forma de administración: Se debe cambiar cada semana el punto de inyección intramuscular. El médico puede prescribir una aguja de 25 mm, de calibre 25 si la considera adecuada para administrar una inyección intramuscular al paciente. En el momento actual, se desconoce durante cuánto tiempo se deben tratar los pacientes. Los pacientes deben ser evaluados clínicamente después de dos años de tratamiento y la continuación del tratamiento debe ser decidida por el médico prescriptor de una forma individualizada. El tratamiento debe ser interrumpido si el paciente desarrolla EM progresiva crónica.
Contraindicaciones.
Inicio del tratamiento en el embarazo (ver sección Fertilidad, embarazo y lactancia). Pacientes con antecedentes de hipersensibilidad al interferón beta recombinante o natural o a alguno de los excipientes incluidos en Composición. Pacientes con depresión severa activa y/o ideación suicida (ver secciones Advertencias y Reacciones adversas).
Reacciones adversas.
La incidencia más alta de reacciones adversas asociadas al interferón beta 1a está relacionada con los síntomas pseudogripales. Los síntomas pseudogripales notificados con más frecuencia son dolores musculares, fiebre, escalofríos, sudoración, astenia, cefalea y náuseas. El ajuste de la dosis de Escleroferon® al inicio del tratamiento ha demostrado una reducción en la gravedad e incidencia de síntomas pseudogripales. Los síntomas pseudogripales tienden a ser más acusados al principio del tratamiento y disminuyen en frecuencia con el tratamiento continuado. Después de las inyecciones pueden aparecer síntomas neurológicos transitorios que recuerden a las exacerbaciones de la EM. En cualquier momento durante el tratamiento pueden aparecer episodios transitorios de hipertonía y/o debilidad muscular intensa que impidan los movimientos voluntarios. Estos episodios tienen una duración limitada, guardan relación temporal con las inyecciones y pueden reaparecer con las siguientes inyecciones. En algunos casos estos síntomas se asocian a síntomas pseudogripales. Las frecuencias de las reacciones adversas se expresan en pacientes-año, de acuerdo con las siguientes categorías: Muy frecuentes (1/10 pacientes-año); Frecuentes (1/100 a < 1/10 pacientes-año); Poco frecuentes (1/1.000 a < 1/100 pacientes-año); Raras (1/10.000 a < 1/1.000 pacientes-año); Muy raras ( < 1/10.000 pacientes-año); Frecuencia no conocida (no puede estimarse a partir de los datos disponibles). La unidad paciente-tiempo es la suma de las unidades individuales de tiempo que el paciente del estudio ha estado expuesto al interferón beta 1a antes de experimentar la reacción adversa. Por ejemplo, 100 personas-año podrían corresponder a una reacción observada en 100 pacientes en tratamiento durante un año o en 200 pacientes tratados durante medio año. En la siguiente tabla se citan las reacciones adversas identificadas en estudios (ensayos clínicos y estudios de observación, con un periodo de seguimiento comprendido entre dos y seis años) y otras reacciones adversas identificadas mediante notificación espontánea tras la comercialización con una frecuencia desconocida. Las reacciones adversas se enumeran en orden decreciente de gravedad dentro de cada intervalo de frecuencia.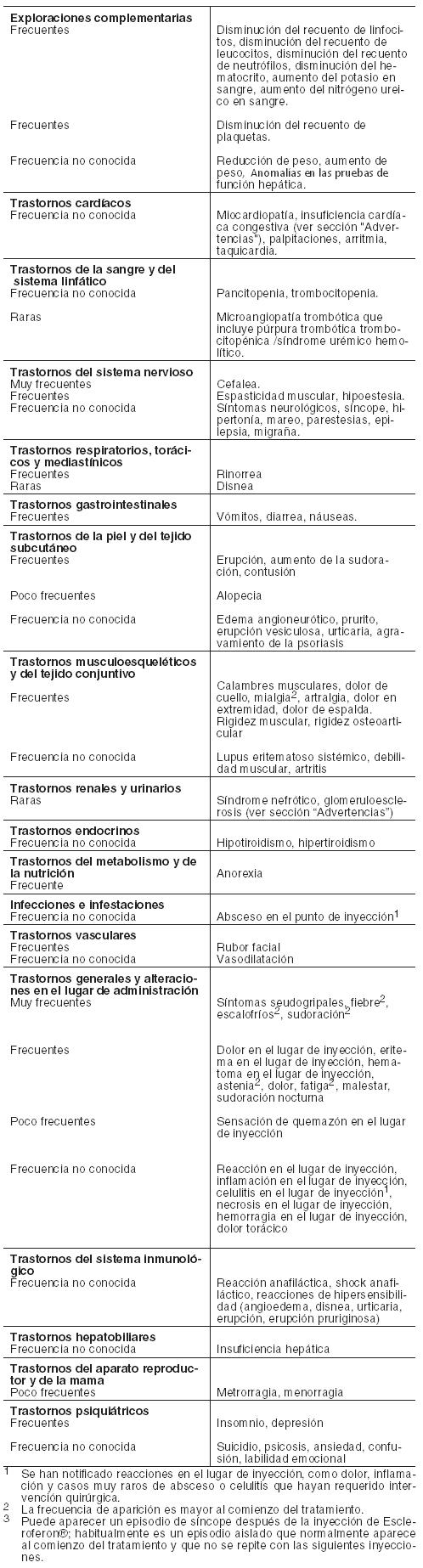
Población pediátrica: Los datos limitados publicados indican que el perfil de seguridad en adolescentes de 12 a 16 años que reciben Escleroferon® 30 microgramos por vía intramuscular una vez a la semana es similar al observado en adultos. Notificación de sospechas de reacciones adversas: Es importante notificar sospechas de reacciones adversas al medicamento tras su autorización. Ello permite una supervisión continua de la relación riesgo/beneficio del medicamento. Se invita a los profesionales sanitarios a notificar las sospechas de reacciones adversas a la Unidad de Seguridad de Medicamentos de BIOSIDUS S.A. Constitución 4234 (C1254ABX) Buenos Aires, Argentina (54-11) 4909-8049. farmacovigilancia@biosidus.com.ar
Advertencias.
Se debe administrar Escleroferon® con precaución a pacientes con trastornos depresivos anteriores o activos, en particular a aquéllos con antecedentes de ideación suicida (ver sección Contraindicaciones). Es sabido que la depresión y la ideación suicida se presentan con mayor frecuencia en la población con esclerosis múltiple y en asociación con el uso de interferón. Se debe aconsejar a los pacientes que informen de inmediato a su médico ante cualquier síntoma de depresión y/o ideas de suicidio. Se debe controlar estrechamente a aquellos pacientes que muestren signos de depresión durante el tratamiento y tratarlos de manera adecuada. Se debe considerar la interrupción del tratamiento con Escleroferon® (ver secciones Contraindicaciones y Reacciones adversas). Se debe administrar Escleroferon® con precaución a pacientes con antecedentes de crisis epilépticas y a los que estén recibiendo tratamiento con antiepilépticos, en particular si la epilepsia no está adecuadamente controlada con antiepilépticos (ver secciones Interacciones y Reacciones adversas). Se debe tener precaución y se debe vigilar estrechamente cuando se administre Escleroferon® a pacientes con insuficiencia renal o hepática grave y a aquellos pacientes con mielosupresión grave. Microangiopatía trombótica (MAT): Se han notificado casos de MAT, manifestada como púrpura trombótica trombocitopénica (PTT) o síndrome urémico hemolítico (SUH), incluidos casos mortales, con medicamentos con interferón beta. Los acontecimientos se notificaron en diferentes momentos del tratamiento y pueden ocurrir transcurridas varias semanas o años después de comenzar el tratamiento con interferón beta. Las manifestaciones clínicas incipientes incluyen trombocitopenia, hipertensión de nueva aparición, fiebre, síntomas del sistema nervioso central (por ejemplo, confusión, paresia) e insuficiencia renal. Los resultados de laboratorio sugestivos de MAT incluyen disminución del recuento de plaquetas, aumento de lactato deshidrogenasa (LDH) en suero debido a la hemólisis y esquistocitos (fragmentación de los eritrocitos) en un frotis de sangre. Por lo tanto, si se observan manifestaciones clínicas de MAT, se recomienda realizar más pruebas para controlar los niveles de plaquetas en sangre, la LDH en suero, frotis de sangre y función renal. Si se diagnostica MAT, es preciso iniciar el tratamiento inmediatamente (considerar recambio plasmático) y se recomienda suspender inmediatamente Escleroferon®. Síndrome nefrótico: se han notificado casos de síndrome nefrótico con diferentes nefropatías subyacentes, incluyendo la glomeruloesclerosis focal y segmentaria colapsante (GESFC), enfermedad con cambios mínimos (ECM), glomerulonefritis membranoproliferativa (GNMP) y glomerulopatía membranosa (GNM) durante el tratamiento con medicamentos a base de interferón beta. Los acontecimientos se notificaron en diversos momentos durante el tratamiento y pueden ocurrir después de varios años de tratamiento con interferón beta. Se recomienda monitorizar periódicamente para detectar signos o síntomas precoces tales como edema, proteinuria y deterioro de la función renal, especialmente en pacientes con mayor riesgo de enfermedad renal. Es necesario tratar inmediatamente el síndrome nefrótico y considerar la suspensión del tratamiento con Escleroferon®. Se ha notificado lesión hepática con niveles elevados de enzimas hepáticas en suero, hepatitis, hepatitis autoinmune e insuficiencia hepática con interferón beta en estudios poscomercialización (ver sección Reacciones adversas). A veces, estas reacciones se han producido en presencia de otros medicamentos asociados a lesión hepática. No se ha determinado la posibilidad de efectos aditivos debidos a varios medicamentos o a otros agentes hepatotóxicos (p. ej., alcohol). Se debe vigilar en los pacientes la aparición de signos de lesión hepática y se tomarán precauciones cuando se utilicen interferones conjuntamente con otros medicamentos asociados a lesión hepática. Se debe controlar estrechamente a aquellos pacientes con enfermedades cardíacas tales como angina, insuficiencia cardíaca congestiva o arritmia por el empeoramiento de su situación clínica durante el tratamiento con Escleroferon®. Los síntomas pseudogripales asociados al tratamiento con interferón beta 1a pueden resultar estresantes para aquellos pacientes con una alteración cardíaca subyacente. El uso de interferón se asocia con alteraciones analíticas. Por lo tanto, además de las pruebas analíticas que se realizan normalmente para controlar a aquellos pacientes con EM, se recomienda llevar a cabo recuento leucocitario completo y diferencial, recuento de plaquetas y química sanguínea, incluyendo pruebas de función hepática durante el tratamiento con Escleroferon®. Aquellos pacientes con mielosupresión pueden necesitar una monitorización más estrecha del recuento hematológico completo, con diferencial y de la cifra de plaquetas. Los pacientes pueden desarrollar anticuerpos frente al interferón beta. Los anticuerpos de algunos de esos pacientes reducen la actividad del interferón beta 1a in vitro (anticuerpos neutralizantes). Los anticuerpos neutralizantes se asocian con una reducción de los efectos biológicos in vivo de Escleroferon® y pueden estar potencialmente asociados con una reducción de la eficacia clínica. Se estima que la meseta de incidencia de formación de anticuerpos neutralizantes se alcanza después de 12 meses de tratamiento. Ensayos clínicos recientes con pacientes tratados con interferón beta 1a hasta tres años sugieren que aproximadamente del 5 % al 8 % desarrolla anticuerpos neutralizantes. La utilización de diversos análisis para detectar los anticuerpos séricos a los interferones limita la capacidad para comparar la antigenicidad entre diferentes productos.
Interacciones.
No se han realizado estudios de interacciones convencionales en humanos. La interacción del interferón beta con corticosteroides o con la corticotropina (ACTH) no ha sido estudiada sistemáticamente. Los ensayos clínicos indican que los pacientes con EM pueden recibir interferón beta 1a y corticosteroides o ACTH durante las recidivas. Se ha comunicado que los interferones reducen la actividad de las enzimas dependientes del citocromo P450 hepático en humanos y animales. Se evaluó el efecto de la administración de interferón beta 1a a alta dosis sobre el metabolismo dependiente de P450 en el mono, y no se observaron cambios en la capacidad de metabolización hepática. Se debe tener precaución cuando se administre Escleroferon® en combinación con otras especialidades farmacéuticas de índice terapéutico estrecho y que dependan en su mayor parte del sistema citocromo P450 hepático para su aclaramiento, tales como antiepilépticos y algunas clases de antidepresivos. Fertilidad, embarazo y lactancia: Embarazo: Los datos relativos al uso de interferón beta 1a durante el embarazo son limitados. Los datos disponibles indican que puede aumentar el riesgo de aborto espontáneo. El inicio del tratamiento durante el embarazo está contraindicado (ver sección Contraindicaciones). Las mujeres en edad fértil deben utilizar métodos anticonceptivos efectivos. Si la paciente queda embarazada o tienen intención de quedarse embarazada durante el tratamiento con Escleroferon®, se le debe informar de los riesgos potenciales y considerar la conveniencia de interrumpir el tratamiento (ver sección Datos preclínicos sobre seguridad). En pacientes con una tasa elevada de recaídas antes del inicio del tratamiento, hay que sopesar el riesgo de una recaída grave después de interrumpir la administración de Escleroferon® en caso de embarazo frente al posible aumento del riesgo de aborto espontáneo. Lactancia: Se desconoce si el interferón beta se excreta en la leche materna. Dada la posibilidad de reacciones adversas graves en los lactantes, se debe decidir si es necesario interrumpir la lactancia o interrumpir el tratamiento con Escleroferon®. Fertilidad: Se han llevado a cabo estudios de fertilidad y desarrollo en el mono rhesus con una forma relacionada del interferón beta 1a. A dosis muy altas, se observaron efectos anovulatorios y abortivos en animales de ensayo (ver sección Datos preclínicos sobre seguridad). No hay datos disponibles sobre los efectos del interferón beta 1a sobre la fertilidad masculina. Efectos sobre la capacidad para conducir y utilizar máquinas: No se han realizado estudios de los efectos del interferón beta sobre la capacidad para conducir y utilizar máquinas. Las reacciones adversas que afectan al sistema nervioso central puede influir ligeramente sobre la capacidad para conducir y utilizar máquinas en pacientes susceptibles (ver sección Reacciones adversas).
Conservación.
Conservar refrigerado (entre 2°C y 8°C). No congelar. Naturaleza y contenido del envase: Jeringa prellenada de 1 ml con capuchón a prueba de manipulaciones y tope de émbolo que contiene 0,5 ml de solución. Tamaño del envase: caja de una, cuatro o doce jeringas prellenadas de 0,5 ml. Cada jeringa está acondicionada en una bandeja de plástico sellada que también contiene una aguja de inyección para vía intramuscular. Precauciones especiales de eliminación y otras manipulaciones: Escleroferon® se presenta listo para usar en una jeringa prellenada para solución inyectable. Una vez retirado del refrigerador, dejar que Escleroferon® en jeringa prellenada adquiera la temperatura ambiente (15°C -25°C) durante 30 minutos aproximadamente. Si la solución inyectable contiene partículas o si tiene cualquier color que no sea claro e incoloro, la jeringa no debe usarse. Se suministra la aguja de inoculación para inyección intramuscular. Cada jeringa prellenada de Escleroferon® contiene una sola dosis única. La porción no utilizada de cualquier jeringa prellenada debe desecharse. La eliminación del medicamento no utilizado y de todos los materiales que hayan estado en contacto con él, se realizará de acuerdo con la normativa local.
Sobredosificación.
No se han notificado casos de sobredosis. Sin embargo, ante la eventualidad de una sobredosificación concurrir al Hospital más cercano o comunicarse con los siguientes Centros especializados: Hospital de Niños Ricardo Gutiérrez (011) 4962-6666. Hospital Posadas: (011) 4654-6648/4658-7777.
Presentación.
Solución inyectable: Escleroferon®: Envases conteniendo 4 jeringas prellenadas de 0,5 ml de solución inyectable, con 4 agujas para inyección intramuscular.
Revisión.
20/02/2017. Disp. N° 1854.