LEVECOM SOLUCION
BALIARDA
Antiepiléptico.
Composición.
Levecom 500: Cada comprimido recubierto ranurado contiene: Levetiracetam 500,00 mg. Excipientes: almidón de maíz, povidona, crospovidona, estearato de magnesio, talco, hidroxipropilmetilcelulosa, dióxido de titanio, polietilenglicol 6000, propilenglicol, dióxido de silicio coloidal, c.s.p. 1 comprimido. Levecom 1000: Cada comprimido recubierto ranurado contiene: Levetiracetam 1000,00 mg. Excipientes: almidón de maíz, povidona, crospovidona, estearato de magnesio, talco, hidroxipropilmetilcelulosa, dióxido de titanio, polietilenglicol 6000, propilenglicol, dióxido de silicio coloidal, óxido férrico amarillo, c.s.p. 1 comprimido. Levecom (solución oral): Cada ml contiene: Levetiracetam 100,00 mg. Excipientes: metilparabeno, propilparabeno, propilenglicol, citrato de sodio dihidrato, ácido cítrico anhidro, sucralosa, sacarina sódica, glicerina, sorbitol 70%, esencia de uva líquida, agua purificada, c.s.p. 1 ml.
Farmacología.
Acción farmacológica: Levetiracetam, es un derivado de la pirrolidona, no relacionado químicamente con otros antiepilépticos existentes. Estudios in vitro e in vivo sugieren que levetiracetam inhibe la descarga epileptiforme sin afectar la excitabilidad neuronal normal, lo cual sugiere que puede prevenir selectivamente la hipersincronización de la descarga epileptiforme y la propagación de la actividad convulsiva. El mecanismo por el cual levetiracetam ejerce su acción terapéutica no ha sido dilucidado. Sin embargo, estudios in vitro revelaron que levetiracetam afecta los niveles cálcicos intraneuronales mediante la inhibición parcial de las corrientes de calcio tipo N y la reducción de la liberación de calcio de la reserva intraneuronal. Asimismo, invierte parcialmente el efecto inhibitorio del zinc y de las b-carbolinas sobre las corrientes dependientes de GABA y glicina. Por otro lado, estudios en tejido cerebral de ratas indican que levetiracetam se une a la proteína 2A de las vesículas sinápticas, la cual participa en la fusión de las vesículas y en la exocitosis de neurotransmisores. Levetiracetam y sus análogos presentan un orden de afinidad por dicha proteína que se correlaciona con la potencia de la actividad anticonvulsivante en el modelo de epilepsia audiogénica en ratón. Este hallazgo sugiere que la interacción de levetiracetam con la proteína 2A de las vesículas sinápticas puede contribuir en el mecanismo de acción del fármaco como antiepiléptico. Farmacocinética: Absorción y Distribución: levetiracetam es rápida y casi completamente absorbido luego de la administración oral. La Cmáx se alcanza, en promedio, luego de una hora de la toma. La ingesta simultánea con alimentos no modifica la extensión de la absorción de levetiracetam pero disminuye un 20% su Cmáx y retrasa 1,5 horas su tmáx. La biodisponibilidad oral de levetiracetam desde la formulación en comprimidos es del 100%, siendo su velocidad y extensión de absorción similares a las de la solución oral. La farmacocinética de levetiracetam es lineal en el rango de dosis entre 500 y 5000 mg y presenta baja variabilidad intra e interindividual. Luego de la administración de dosis múltiples, el estado estacionario se alcanza en el lapso de aproximadamente 2 días. Levetiracetam y su principal metabolito presentan una baja unión a proteínas plasmáticas ( < 10%). Metabolismo: levetiracetam no se metaboliza extensamente en humanos. El principal metabolito, producto de la hidrólisis enzimática del grupo acetamida, es inactivo y no es dependiente del citocromo P450. Eliminación: el t½ en adultos es de 7 ± 1 horas. El clearance total es de 0,96 ml/min/kg y el renal es de 0,6 ml/min/kg. El 66% de la dosis administrada se elimina en la orina, principalmente como droga sin metabolizar. El mecanismo de excreción es filtración glomerular seguida de reabsorción tubular parcial. El clearance de levetiracetam se correlaciona con el de creatinina. Poblaciones especiales: Pacientes de edad avanzada: luego de la administración de levetiracetam durante 10 días en pacientes de edad avanzada, el clearance total disminuyó un 38% y el t1/2 fue 2,5 horas más prolongado que el de los pacientes jóvenes. Estos hallazgos pueden atribuirse a la disminución de la función renal observada en este grupo etario. Pacientes pediátricos: luego de la administración de una dosis oral única (20 mg/kg) en niños de 6 a 12 años de edad, se observó un incremento del 40% en el clearance aparente ajustado por el peso corporal, en comparación con pacientes adultos. Tras la administración de dosis repetidas (20, 40 y 60 mg/kg/día) en niños de 4-12 años de edad, levetiracetam se absorbió rápidamente. La Cmáx se alcanzó a la hora de la toma y el t½ fue de 5 horas. La farmacocinética fue lineal dentro del rango de dosis administrado. Tras la administración de una dosis única (20 mg/kg) de solución oral en niños con epilepsia (1 mes a < 4 años de edad), levetiracetam se absorbió rápidamente. La Cmáx se alcanzó a la hora de la toma, el t½ fue de 5 horas y el clearance aparente fue de 1,5 ml/min/kg. El análisis farmacocinético poblacional mostró una alta correlación entre el peso corporal y el clearance de levetiracetam en pacientes pediátricos; el clearance se incrementó con el incremento del peso corporal. Insuficiencia renal: el clearance total de levetiracetam se redujo respectivamente un 40%, 50% y 60% en pacientes con insuficiencia renal leve (Clcr 50-80 ml/min), moderada (Clcr 30-50 ml/min) y severa (Clcr < 30 ml/min) comparado con individuos con función renal normal. Luego de una sesión de hemodiálisis de 4 horas, la concentración de levetiracetam se reduce aproximadamente un 50%. Insuficiencia hepática: en pacientes con insuficiencia hepática leve (Child-Pugh A) a moderada (Child-Pugh B) no se observaron cambios en la farmacocinética de levetiracetam. En pacientes con insuficiencia hepática severa (Child-Pugh C), el clearance total se redujo en un 50% en comparación con sujetos con función hepática normal, pero la disminución del clearance renal representa la mayor parte de esta reducción.
Indicaciones.
Como monoterapia en el tratamiento de las crisis de inicio parcial con o sin generalización secundaria en adultos y adolescentes mayores de 16 años con un diagnóstico reciente de epilepsia. Tratamiento adyuvante de las crisis de inicio parcial con o sin generalización secundaria en adultos, adolescentes y niños de 4 años o más con epilepsia. Tratamiento adyuvante de las crisis mioclónicas en adultos y adolescentes de 12 años o más con Epilepsia Mioclónica Juvenil. Tratamiento adyuvante de las crisis tónico-clónicas generalizadas primarias en adultos y niños de 6 años o más con Epilepsia Generalizada Idiopática.
Dosificación.
Crisis de inicio parcial con o sin generalización secundaria: Como monoterapia: Adultos y adolescentes ≥16 años: Dosis inicial: 250 mg dos veces/día. Luego de dos semanas de tratamiento, la dosis debería ser incrementada a 500 mg dos veces/día. En función de la respuesta clínica, cada dos semanas la dosis podrá incrementarse a razón de 500 mg/día. Dosis máxima recomendada: 1500 mg dos veces/día. Como tratamiento adyuvante: Adultos y adolescentes ≥16 años: Dosis inicial: 500 mg dos veces/día. Cada dos semanas la dosis puede ser incrementada a razón de 1000 mg/día. Dosis máxima recomendada: 1500 mg dos veces/día. No se ha evidenciado un beneficio adicional con dosis superiores a 3000 mg/día. Niños de 4 a 16 años: Dosis inicial: 10 mg/kg dos veces/día. Cada dos semanas la dosis debería ser incrementada a razón de 20 mg/kg/día. Dosis recomendada: 30 mg/kg dos veces/día. Si el paciente no puede tolerar esta dosis, la dosis diaria puede reducirse. La dosis máxima es de 3000 mg/día. Esquema posológico orientativo para el uso de comprimidos en niños: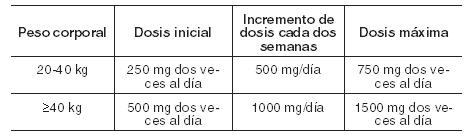
Crisis mioclónicas en pacientes con epilepsia mioclónica juvenil: Adultos y niños ≥12 años: Dosis inicial: 500 mg dos veces/día. Cada dos semanas la dosis debería ser incrementada a razón de 1000 mg/día. Dosis recomendada: 1500 mg dos veces/día. Crisis tónico-clónicas generalizadas primarias: Adultos y adolescentes ≥16 años: Dosis inicial: 500 mg dos veces/día. Cada dos semanas la dosis debería ser incrementada a razón de 1000 mg/día. Dosis recomendada: 1500 mg dos veces/día. Pacientes pediátricos de 6 a 16 años: Dosis inicial: 10 mg/kg dos veces/día. Cada dos semanas la dosis debería ser incrementada a razón de 20 mg/kg/día. Dosis recomendada: 30 mg/kg dos veces/día. En pacientes con peso corporal ≤20 kg se recomienda el empleo de solución oral. Poblaciones especiales: Insuficiencia hepática: no se requiere un ajuste de la dosis. Insuficiencia renal: la siguiente tabla presenta las dosis sugeridas en pacientes con alteración de la función renal: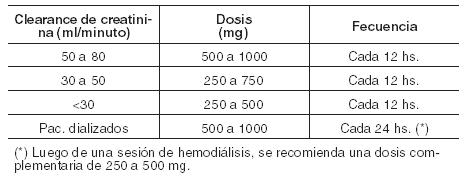
El clearance de creatinina puede ser estimado usando la ecuación de Cockcroft y Gault:
Discontinuación del tratamiento: se recomienda una discontinuación gradual del tratamiento (ej. en adultos y adolescentes con peso corporal ≥50 kg, reducciones de 1000 mg/día cada 2-4 semanas; en niños mayores de 6 meses y adolescentes con peso corporal ≤50 kg, las reducciones de dosis no deberían exceder de los 20 mg/kg/día, cada dos semanas; en niños menores de 6 meses, las reducciones de dosis no deberían exceder de los 14 mg/kg/día, cada dos semanas). Modo de administración: El producto puede administrarse con o fuera de las comidas. La solución oral puede diluirse en agua. Administración de la solución oral con jeringa dosificadora: 1. Retirar el inserto perforado del extremo de la jeringa dosificadora. 2. Insertar el mismo en la boca del frasco y presionar hasta que calce perfectamente. 3. Introducir la jeringa dosificadora en el orificio del inserto perforado ya colocado. 4. Invertir el frasco y aspirar la solución tirando del émbolo lentamente hasta la marca correspondiente al volumen y/o peso que se desee administrar. 5. Retirar la jeringa dosificadora del inserto. 6. Descargar la solución en la boca del paciente, deslizando el émbolo hacia abajo. 7. Cerrar el frasco sin retirar el inserto perforado. 8. Separar los componentes de la jeringa y lavarlos con abundante agua fría, de manera que queden aptos para la próxima utilización.
Contraindicaciones.
Hipersensibilidad a levetiracetam, a otros derivados de la pirrolidona, o a cualquiera de los componentes del producto. Las reacciones de hipersensibilidad pueden incluir anafilaxia y angioedema (ver Advertencias).
Reacciones adversas.
Crisis de inicio parcial: Adultos: las reacciones adversas más comúnmente observadas en estudios clínicos controlados en pacientes adultos tratados con levetiracetam en combinación con otras DAEs y con una incidencia superior a placebo, fueron: infección, astenia, somnolencia y mareos. Estas tres últimas se presentaron principalmente durante las primeras cuatro semanas de tratamiento con levetiracetam. Otras reacciones observadas con una incidencia ≥1% en pacientes adultos tratados con levetiracetam en combinación con otras DAEs y con una incidencia superior a placebo, fueron: Gastrointestinales: anorexia. Neurológicas y psiquiátricas: depresión, nerviosismo, ataxia, vértigo, amnesia, ansiedad, hostilidad, parestesia, labilidad emocional. Respiratorias: faringitis, rinitis, tos incrementada, sinusitis. Sensoriales: diplopía. Otras: cefalea, dolor. Reacciones adversas asociadas con la discontinuación del tratamiento o reducción de la dosis: aproximadamente un 15% de los pacientes adultos tratados con levetiracetam en estudios clínicos discontinuaron el tratamiento o disminuyeron la dosis debido a las reacciones adversas, mientras que el porcentaje de pacientes tratados con placebo que abandonaron el tratamiento fue de 12%. Estas incluyen mareos (1% vs 0% del grupo tratado con placebo) y somnolencia (4% vs 2% del grupo tratado con placebo). Pacientes pediátricos de 4 a 16 años: las reacciones adversas comúnmente observadas en estudios clínicos controlados contra placebo en niños (4-16 años) tratados con levetiracetam en combinación con otras DAEs y con una incidencia superior a placebo, fueron: fatiga, agresión, congestión nasal, disminución del apetito, irritabilidad. Otras reacciones observadas con una incidencia ≥2% en pacientes pediátricos tratados con levetiracetam en combinación con otras DAEs y con una incidencia superior a placebo, fueron: Gastrointestinales: vómitos, dolor abdominal superior, diarrea, gastroenteritis, constipación. Neurológicas: mareos, cefalea, somnolencia, letargo, sedación. Psiquiátricas: labilidad afectiva, agitación, depresión, confusión, comportamiento anormal, insomnio, alteraciones del estado de ánimo, ansiedad, cambios en el humor. Respiratorias: rinitis, tos, nasofaringitis, dolor faringolaríngeo. Sensoriales: conjuntivitis, dolor de oído. Músculo-esqueléticas: artralgia, dolor de cuello. Otras: lesión en la cabeza, injuria, caídas, esguinces, anorexia, gripe. Reacciones adversas asociadas con la discontinuación del tratamiento o reducción de la dosis: aproximadamente un 7% de los pacientes pediátricos tratados con levetiracetam en estudios clínicos discontinuaron el tratamiento debido a las reacciones adversas. Pacientes pediátricos de 1 mes a 4 años: las reacciones adversas comúnmente observadas en estudios clínicos controlados contra placebo en niños (1 mes-4 años) tratados con levetiracetam en combinación con otras DAEs y observados con una incidencia superior a placebo, fueron: somnolencia, irritabilidad. Aproximadamente un 3% de los pacientes pediátricos tratados con levetiracetam en estudios clínicos discontinuaron el tratamiento o fue necesaria una reducción de la dosis debido a somnolencia (13%) e irritabilidad (12%), en comparación con el grupo placebo (2% y 0%, respectivamente). Convulsiones mioclónicas: Las reacciones adversas más comúnmente observadas en estudios clínicos controlados contra placebo en adultos y adolescentes (≥12 años) tratados con levetiracetam en combinación con otras DAEs y observados con una incidencia superior a placebo, fueron: somnolencia, dolor de cuello y faringitis. Otras reacciones observadas con una incidencia ≥5% en pacientes ≥ de 12 años tratados con levetiracetam en combinación con otras DAEs y con una incidencia superior a placebo, fueron: Sensoriales: vértigo. Infecciosas: gripe. Psiquiátricas: depresión. Aproximadamente un 8% de los pacientes tratados con levetiracetam en estudios clínicos discontinuaron el tratamiento debido a las reacciones adversas. Estas incluyen: ansiedad, humor depresivo, depresión, diplopía, hipersomnia, insomnio, irritabilidad, nerviosismo, somnolencia. Crisis tónico-clónicas generalizadas primarias: La reacción adversa más comúnmente observada en estudios clínicos controlados contra placebo en adultos y niños mayores de 4 años tratados con levetiracetam en combinación con otras DAEs y con una incidencia superior a placebo, fue nasofaringitis. Otras reacciones observadas con una incidencia ≥5% en pacientes tratados con levetiracetam en combinación con otras DAEs y con una incidencia superior a placebo, fueron: Gastrointestinales: diarrea. Psiquiátricas: irritabilidad, cambios en el humor. Respiratorias: nasofaringitis. Otras: fatiga. Las reacciones adversas asociadas con la discontinuación del tratamiento afectaron al 5% de los pacientes tratados con levetiracetam en estudios clínicos controlados, comparado con el 8% de los pacientes del grupo placebo. Reportes postcomercialización: otras reacciones adversas reportadas con levetiracetam (relación causal desconocida) incluyen: parámetros anormales de la función hepática, falla renal aguda, anafilaxia, angioedema, agranulocitosis, coreoatetosis, reacción cutánea con eosinofilia y síntomas sistémicos (DRESS), disquinesia, eritema multiforme, insuficiencia hepática, hepatitis, hiponatremia, debilidad muscular, rabdomiolisis (significativamente mayor en pacientes japoneses), pancreatitis, pancitopenia (con supresión de médula ósea en algunos casos), ataque de pánico, trombocitopenia, pérdida de peso, alopecia.
Precauciones.
Reacciones dermatológicas serias: se reportaron reacciones dermatológicas serias, incluyendo síndrome de Stevens-Johnson y necrólisis epidérmica tóxica en pacientes adultos y pediátricos tratados con levetiracetam. Estos efectos fueron reportados en promedio luego de 14 a 17 días de iniciado el tratamiento con levetiracetam, aunque han habido reportes aún luego de 4 meses de iniciado el tratamiento. En caso de un primer signo de erupción, se recomienda suspender el tratamiento a menos que la misma sea claramente no relacionada con levetiracetam. Anormalidades hematológicas: en estudios clínicos se observaron alteraciones hematológicas, como disminución en el recuento de los glóbulos rojos, recuento de glóbulos blancos, neutrófilos, hemoglobina y hematocrito; y aumento de eosinófilos. Durante la postcomercialización se han reportado casos de agranulocitosis, pancitopenia y trombocitopenia. Se recomienda realizar un análisis completo de sangre en pacientes que experimentan debilidad, pirexia, infecciones recurrentes o desórdenes de coagulación. En estudios clínicos controlados en pacientes adultos con crisis de inicio parcial, se reportaron disminuciones leves pero estadísticamente significativas en el recuento de glóbulos rojos (0,03 x 106/mm3), hematocrito promedio (0,38 %) y hemoglobina promedio (0,09 g/dl) en el grupo tratado con levetiracetam, en comparación con el grupo placebo. Asimismo se reportaron, con una incidencia superior a placebo, disminuciones posiblemente significativas de los recuentos de glóbulos blancos (≤2,8 x 109/l) y de neutrófilos (≤1 x 109/l). En estudios clínicos controlados en pacientes pediátricos de 4 a 16 años con crisis de inicio parcial, se reportaron disminuciones leves pero estadísticamente significativas en los recuentos de glóbulos blancos y de neutrófilos en el grupo tratado con levetiracetam, en comparación con el grupo placebo. Asimismo se reportó un incremento en el recuento linfocitario relativo promedio en el 1,7% de los pacientes tratados con levetiracetam, en comparación con un descenso del mismo observado en el 4% del grupo placebo; y un incremento clínicamente significativo en el recuento de eosinófilos (8,6% en el grupo tratado con levetiracetam vs 6,1% en el grupo placebo). Dado que el número de pacientes con Epilepsia Mioclónica Juvenil tratados con levetiracetam fue considerablemente menor al número de pacientes con crisis de inicio parcial, debe considerarse que los valores observados en estos últimos puedan también presentarse en pacientes con Epilepsia Mioclónica Juvenil. Incremento en la presión sanguínea: en estudios clínicos controlados en pacientes pediátricos de 1 mes a 4 años de edad, se observaron mediciones aisladas de la presión arterial diastólica significativamente incrementadas en el grupo tratado con levetiracetam (17%), en comparación con el grupo placebo (2%). No se encontraron diferencias en la presión arterial diastólica promedio en ambos grupos. Esta disparidad con respecto a placebo no se observó en otros grupos etarios. Se recomienda controlar a los pacientes de 1 mes a 4 años, debido a los aumentos de la presión arterial diastólica. Control de las convulsiones durante el embarazo: los cambios fisiológicos durante el embarazo, principalmente en el tercer trimestre, pueden disminuir gradualmente los niveles de levetiracetam. Por lo tanto, se recomienda monitorear cuidadosamente a las pacientes durante el embarazo y continuar durante el período postparto si durante el embarazo hubo cambios en la dosis. Poblaciones especiales: Insuficiencia renal: ver Dosificación. Insuficiencia hepática: en pacientes con insuficiencia hepática severa, se recomienda la valoración de la función renal antes de iniciar el tratamiento con levetiracetam. Pacientes de edad avanzada: los estudios clínicos no revelaron diferencias significativas en la seguridad entre los pacientes mayores y menores de 65 años. Levetiracetam se excreta principalmente por vía renal, y el riesgo de padecer reacciones adversas es mayor en pacientes con insuficiencia renal. Dado que los pacientes de edad avanzada son más propensos a sufrir trastornos de la función renal, se recomienda precaución durante la selección de la dosis, sugiriéndose el monitoreo de la función renal. Pacientes pediátricos: levetiracetam solución oral es la formulación más adecuada para administrar en lactantes y niños con un peso de 25 kg o inferior. Embarazo: durante la experimentación en ratas y conejos con dosis iguales o superiores a la dosis máxima recomendada en humanos, se observó un incremento en la incidencia de malformaciones fetales (incluyendo anormalidades esqueléticas fetales leves), retardo en el crecimiento pre y/o postnatal, reducción del peso corporal fetal y un incremento en la mortalidad embriofetal y de las crías. La discontinuación de los antiepilépticos puede dar lugar a una exacerbación de la enfermedad, que podría perjudicar a la madre y al feto. No habiendo estudios adecuados con levetiracetam en mujeres embarazadas, el producto debería ser usado durante el embarazo sólo si los beneficios potenciales superan los posibles riesgos para el feto. Lactancia: levetiracetam se excreta en la leche humana. En consecuencia, la decisión de discontinuar el tratamiento o la lactancia deberá ser tomada de acuerdo con la importancia que la droga posea para la madre. Interacciones medicamentosas: Dado que levetiracetam no inhibe ni es sustrato de las distintas isoformas del citocromo P450, epóxido hidrolasa o UDP-glucuronosiltransferasa y no se une a proteínas plasmáticas en forma apreciable, es improbable que produzca interacciones farmacocinéticas o sea susceptible a ellas. Otros antiepilépticos: levetiracetam no modifica las concentraciones plasmáticas de las siguientes DAEs inductoras de enzimas: fenitoína, ácido valproico (valproato), carbamazepina, gabapentin, lamotrigina, fenobarbital y primidona. Asimismo no se observaron cambios relevantes en la farmacocinética de levetiracetam luego de la administración concomitante con dichas drogas. Sin embargo, en estudios clínicos en pacientes pediátricos se observó un incremento del 22% en el clearance de levetiracetam en niños tratados concomitantemente con DAEs inductoras de enzimas. No obstante, no se requiere un ajuste de dosis durante el tratamiento concomitante de levetiracetam y dichos antiepilépticos. Anticonceptivos: la administración de 500 mg dos veces al día de levetiracetam no afectó la farmacocinética de un anticonceptivo conteniendo 0,03 mg de etinilestradiol y 0,15 mg de levonorgestrel ni los niveles de hormona luteinizante ni progesterona. Asimismo no se observaron cambios relevantes en la farmacocinética de levetiracetam luego de la coadministración con dicho anticonceptivo. Digoxina: la administración de 1000 mg dos veces al día de levetiracetam no modificó la farmacocinética ni la farmacodinamia de digoxina (0,25 mg/día). La coadministración de digoxina no modificó la farmacocinética de levetiracetam. Warfarina: la administración concomitante de 1000 mg dos veces al día de levetiracetam y warfarina no modificó la farmacocinética de ninguna de las dos drogas ni alteró el tiempo de protrombina. Probenecid: tras la administración concomitante de probenecid (500 mg cuatro veces al día), un inhibidor de la secreción tubular renal, y levetiracetam (1000 mg dos veces al día), la Cmáx en el estado estacionario del principal metabolito se duplicó mientras que su clearance renal disminuyó un 60%, probablemente debido a la inhibición competitiva de la secreción tubular del principal metabolito. No se observaron cambios en la farmacocinética de levetiracetam ni en la fracción de droga sin metabolizar excretada por vía renal. Es de esperar que otras drogas que se excretan por secreción tubular activa puedan reducir asimismo el clearance renal del principal metabolito de levetiracetam. No se ha estudiado el efecto de levetiracetam sobre probenecid u otros fármacos secretados activamente (como AINES, sulfonamidas). Metotrexato: luego de la administración concomitante de levetiracetam y metotrexato, se produce un aumento de la Cmáx de metotrexato a niveles potencialmente tóxicos debido a la disminución del clearance de metotrexato.
Advertencias.
Alteraciones en el comportamiento y síntomas psicóticos: Alteraciones en el comportamiento: en estudios clínicos en pacientes adultos y pediátricos con crisis mioclónicas y tónico-clónicas generalizadas primarias, la incidencia de anormalidades en el comportamiento fue comparable a la de los estudios en pacientes con crisis de inicio parcial. Durante la evaluación de pacientes adultos y pediátricos (4 a 16 años) se reportaron, con una incidencia superior a placebo, trastornos tales como: agresión, agitación, ira, ansiedad, apatía, despersonalización, depresión, labilidad emocional, hostilidad, hiperkinesia, irritabilidad, nerviosismo, neurosis y desórdenes de personalidad. En pacientes pediátricos de 1 mes a < 4 años, la administración de levetiracetam se asoció con irritabilidad. Un total de 1,7% de los pacientes adultos tratados y un 0,2% de los pacientes que recibieron placebo, discontinuaron el tratamiento debido a estos eventos adversos. En pacientes pediátricos, un 11% de los pacientes tratados y un 6% de los pacientes que recibieron placebo discontinuaron el tratamiento. Síntomas psicóticos: en estudios clínicos, el 1% de los pacientes adultos, el 2% de los pacientes pediátricos (4 a 16 años) y el 17% de los pacientes pediátricos (1 mes a < 4 años) tratados con levetiracetam, experimentaron síntomas psicóticos en comparación con el grupo placebo, siendo los valores 0,2%, 0,2% y 5%; respectivamente. En un estudio controlado se evaluaron los efectos neurocognitivos y conductuales en pacientes pediátricos (4 a 16 años) tratados con levetiracetam. El síntoma reportado fue paranoia (1,6% vs 0% del grupo tratado con placebo). Asimismo se reportó confusión en el 3,1% de los pacientes tratados con levetiracetam (vs 0% del grupo tratado con placebo). En estudios clínicos, se reportaron 2 casos de psicosis en pacientes adultos tratados con levetiracetam, que debieron ser hospitalizados y discontinuar el tratamiento. En ambos casos, el evento se observó dentro de la primera semana de iniciado el tratamiento y se resolvieron entra la primera y segunda semana de discontinuado el mismo. Ideación y comportamiento suicida: las drogas antiepilépticas (DAEs), incluyendo levetiracetam, pueden aumentar el riesgo de ideación o de comportamiento suicida en los pacientes cualquiera sea la indicación para la cual han sido prescriptas. Estos pacientes deben ser supervisados ante la posible aparición o el empeoramiento de una depresión preexistente, pensamientos o comportamientos suicidas, y/o ante cualquier cambio inusual en el humor o el comportamiento. En la evaluación de 199 estudios clínicos controlados sobre 11 DAEs utilizadas para tratar epilepsia, trastorno bipolar, migraña y dolor neuropático (meta-análisis realizado por la FDA en 2008), se detectó que los pacientes que recibieron estas drogas tuvieron el doble de riesgo de comportamiento o ideación suicida que aquellos que recibieron placebo (Riesgo Relativo ajustado 1.8, IC 95%: 1.2, 2.7). En los estudios hubo cuatro suicidios en pacientes tratados con DAEs y ninguno en los pacientes tratados con placebo, pero este número es demasiado pequeño para permitir extraer alguna conclusión sobre el efecto de las DAEs sobre el suicidio. El médico que considere prescribir cualquier DAE debe balancear este riesgo con el de la enfermedad no tratada. Las indicaciones para las cuales se prescriben DAEs comprenden patologías que en sí mismas se asocian a un riesgo creciente de morbi-mortalidad y de ideación y comportamiento suicida. Los pacientes, sus cuidadores, y las familias deben ser informados del aumento de riesgo de ideas y comportamientos suicidas causados por DAEs, y se les debe advertir sobre la necesidad de estar alerta ante la aparición o el empeoramiento de los síntomas de depresión, cualquier cambio inusual en el humor o el comportamiento, o la aparición de ideas y comportamiento suicidas. Somnolencia y fatiga: en estudios en pacientes adultos y pediátricos con crisis mioclónicas y tónico-clónicas generalizadas primarias y en pacientes pediátricos con crisis de inicio parcial, la incidencia de somnolencia y fatiga fue comparable a la de los estudios en pacientes adultos con crisis de inicio parcial. En estudios clínicos de pacientes adultos con epilepsia que experimentan convulsiones de inicio parcial, en el 15% de los pacientes tratados con levetiracetam se observó somnolencia y astenia, comparado con el 8% y el 9%, respectivamente, de los pacientes tratados con placebo. Mientras que el 0,8% de los pacientes tratados con levetiracetam, suspendió el tratamiento debido a astenia, en comparación con el 0,5% de los pacientes tratados con placebo. Un 3% de los pacientes tratados y un 0,7% de los pacientes que recibieron placebo discontinuaron el tratamiento debido a somnolencia. La aparición de somnolencia y astenia se observó más frecuentemente durante las primeras cuatro semanas de tratamiento. Debido a la posible aparición de somnolencia y fatiga, especialmente al inicio del tratamiento o después de un incremento de la dosis, se debe monitorear a los pacientes en busca de estos síntomas y los pacientes deberán abstenerse de operar maquinarias peligrosas o conducir automóviles, hasta que conozcan su susceptibilidad personal al fármaco. Anafilaxia y angioedema: levetiracetam puede causar anafilaxia o angioedema luego de la primera dosis o durante el tratamiento. Los signos y síntomas reportados durante la etapa postcomercialización incluyeron hipotensión, urticaria, rash, distrés respiratorio e hinchazón de cara, labios, boca, ojos, lengua, garganta y pies. En algunos casos las reacciones fueron amenazantes para la vida y requirieron tratamiento de emergencia. Se debe advertir a los pacientes, que en caso de que se presenten signos o síntomas de anafilaxia o angioedema, el tratamiento con levetiracetam debe ser discontinuado y debe buscar atención médica inmediata. Dificultades en la coordinación: en estudios de pacientes adultos con crisis de inicio parcial se ha reportado, con una incidencia superior a placebo, dificultades en la coordinación (ataxia, marcha anormal, incoordinación). Un 0,4% de los pacientes tratados (vs 0% del grupo placebo), discontinuaron el tratamiento debido a estos eventos adversos. Dichos eventos ocurrieron más frecuentemente durante las primeras cuatro semanas de tratamiento. Lesión renal aguda: raramente se ha asociado el uso de levetiracetam con lesión renal aguda, siendo el tiempo de aparición de días hasta meses. Discontinuación del tratamiento: levetiracetam debe ser discontinuado en forma gradual para minimizar la posibilidad de un aumento de la frecuencia convulsiva (ver Dosificación).
Conservación.
Levecom 500 - 1000 (comprimidos recubiertos ranurados): Mantener a temperatura no superior a 30°C. Proteger de la humedad. Levecom (solución oral): Mantener a temperatura no superior a 30°C. Proteger de la luz. Una vez abierto, puede utilizarse durante un máximo de 2 meses.
Sobredosificación.
Se han reportado sobredosis que involucran cantidades de hasta 6000 mg/día de levetiracetam. Los signos y síntomas incluyen somnolencia, agitación, agresión, niveles de conciencia deprimidos, depresión respiratoria y coma. Tratamiento: no existe antídoto específico para levetiracetam. Se recomiendan medidas de soporte generales, incluyendo monitoreo frecuente de los signos vitales y observación del paciente. Si está indicado, se puede inducir la emesis o realizar un lavado gástrico; se deberán tomar las medidas necesarias para proteger la ventilación. Aunque se carece de experiencia clínica en el tratamiento de sobredosis, levetiracetam se elimina eficazmente del plasma mediante hemodiálisis. Ante la eventualidad de una sobredosificación, concurrir al Hospital más cercano o consultar a los Centros Toxicológicos de: Hospital Posadas (011) 4654-6648 / 4658-7777. Hospital de Pediatría Ricardo Gutiérrez (011) 4962-2247 / 6666.
Presentación.
Levecom 500: Envase con 20, 30 y 60 comprimidos recubiertos ranurados. Levecom 1000: Envase con 30 y 60 comprimidos recubiertos ranurados. Levecom (solución oral): Frasco conteniendo 300 ml acompañado de jeringa plástica graduada e inserto de polietileno.