Cotellic®
ROCHE
Cotellic es un fármaco de molécula pequeña que produce una inhibición dirigida potente y muy selectiva de las tirosina-treonina-quinasas MEK1 y MEK2.
Composición.
Cada comprimido recubierto contiene 22,20 mg de hemifumarato de cobimetinib (20 mg de cobimetinib base libre), en un excipiente compuesto por: Masa del núcleo del comprimido: Celulosa microcristalina 54,72 mg, lactosa monohidratada 36,48 mg, croscarmelosa sódica 4,80 mg y estearato de magnesio 1,80 mg. Cuerpo de la cubierta del comprimido: Poli (alcohol vinílico) 1,92 mg, dióxido de titanio 1,20 mg, Macrogol/PEG 3350: 0,97 mg, talco 0,71 mg y agua purificada (no corresponde, ya que este disolvente se elimina durante el procesamiento).
Farmacología.
Código ATC: L01XE38. Grupo farmacoterapéutico: Agente antineoplásico, inhibidor de la proteína quinasa. Propiedades farmacodinámicas: Mecanismo de acción: La vía de señalización de la proteína quinasa activada por mitógenos (MAPK) / quinasa regulada por señal extracelular (MEK) es clave para regular la proliferación celular, la regulación del ciclo celular, la supervivencia celular, la angiogénesis y la migración de células. Cobimetinib es un inhibidor reversible, selectivo, alostérico y oral que bloquea la ruta de las proteínquinasas activadas por mitógenos (MAPK) dirigiéndose a la quinasa activada por mitógenos reguladora de la señal extracelular (MEK)1 y MEK2, lo que provoca una inhibición de la fosforilación de la quinasa reguladora de la señal extracelular (ERK)1 y ERK2. La mediación funcional de la vía MAPK es dependiente de la actividad de ERK 1 y 2 que fosforila a las proteínas objetivo en el citoplasma y en el núcleo que inducen la progresión del ciclo celular, la proliferación celular, la supervivencia y la migración. Se ha demostrado una alta potencia inhibitoria en ensayos celulares y bioquímicos, así como la amplia actividad antitumoral in vivo en modelos de xenoinjertos tumorales, incluyendo aquellos mutados por BRAF y KRAS. Por lo tanto, cobimetinib bloquea la actividad pro-mitogénica y oncogénica inducida por la ruta de la MAPK mediante la inhibición de la señalización a nivel de MEK1/2. En estudios bioquímicos y estructurales, se ha demostrado que Cotellic interactúa con MEK de una manera en la que es menos susceptible a los cambios conformacionales dinámicos observados con el estado de fosforilación de MEK. Como resultado Cotellic mantiene la afinidad de la unión y la actividad inhibidora cuando MEK se fosforila. Debido a este diferente mecanismo alostérico de la inhibición, Cotellic ha demostrado la actividad más fuerte en líneas celulares de cáncer y tumores con niveles altos de MEK fosforilado, como se observa frecuentemente en tumores con BRAF mutado. En los modelos preclínicos, la combinación de cobimetinib (inhibidor MEK 1 y 2) y Vemurafenib (inhibidor BRAF) en células de melanoma, demostró que el bloqueo dual impide la reactivación de la vía MAPK provocando una fuerte inhibición de la señalización intracelular lo que conlleva a una mejor respuesta tumoral y una disminución de la proliferación de las células tumorales. Eficacia clínica y seguridad: No se dispone de datos sobre la seguridad o eficacia de Cotellic en combinación con vemurafenib en pacientes con metástasis de sistema nervioso central o en aquéllos con melanoma maligno no-cutáneo. Estudio GO28141 (coBRIM): Es un ensayo clínico multicéntrico, aleatorizado, doble-ciego, controlado con placebo, Fase III, que evaluó la seguridad y eficacia de Cotellic en combinación con vemurafenib, en comparación con vemurafenib más placebo, en pacientes con mutación BRAF V600 positiva que no habían sido previamente tratados y que padecían melanoma no resecable localmente avanzado (estadio IIIc) o melanoma metastásico (estadio IV). En el estudio GO28141 solamente se incluyeron pacientes con un estado de desarrollo ECOG de 0 y 1. Se excluyeron del mismo los que se hallaban en un estado de desarrollo ECOG de 2 o superior. Después de la confirmación de la mutación BRAF V600, mediante el Test de Cobas® 4800 para la mutación BRAF V600, se aleatorizaron a 495 pacientes no tratados previamente con melanoma no resecable localmente avanzado o melanoma metastásico para recibir: Placebo una vez por día, los días 1-21 de cada ciclo de tratamiento de 28 días y 960 mg de vemurafenib dos veces por día, los días 1-28, o bien Cotellic 60 mg una vez por día, los días 1-21 de cada ciclo de tratamiento de 28 días y 960 mg de vemurafenib dos veces por día, los días 1-28. La variable principal evaluada por el Investigador fue la sobrevida libre de progresión (SLP). Las variables secundarias de eficacia incluyeron la sobrevida global (SG), la tasa de respuesta objetiva (TRO), la duración de la respuesta (DdR) evaluadas por el Investigador y la SLP por un Comité de Revisión Independiente (CRI). Las principales características basales incluyeron: el 58% de los pacientes eran varones, la edad mediana de 55 años (rango de 23 a 88 años), el 60% tenía melanoma metastásico en estadio M1c y la proporción de pacientes con LDH elevado del 46,3% en el grupo tratado con cobimetinib más vemurafenib y del 43,0% en el que recibió placebo más vemurafenib. En el estudio GO28141 había 89 pacientes (18,1%) de 65-74 años, 38 (7,7%) de 75-84 años y 5 (1,0%) de 85 años o mayores. Los resultados de eficacia se resumen en la Tabla 1.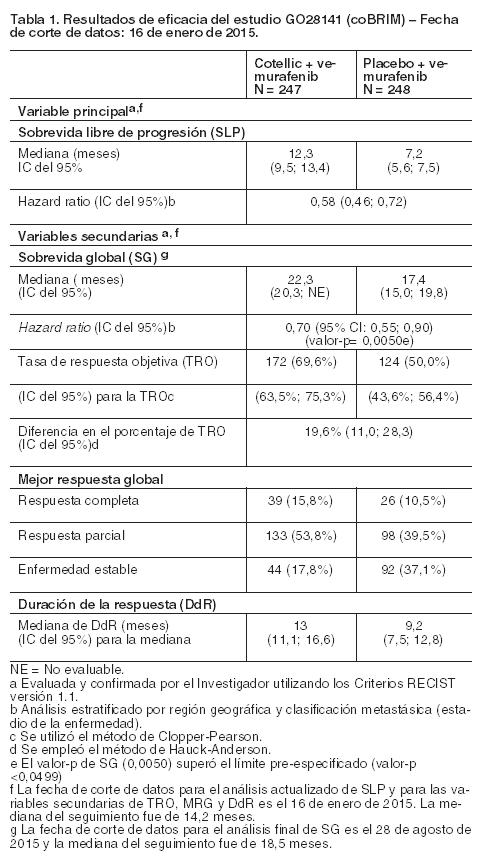
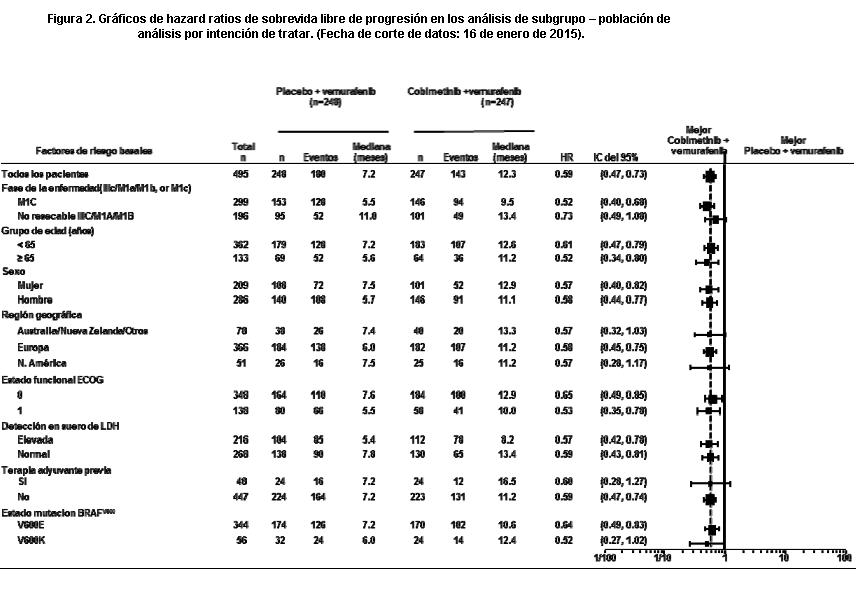
Los análisis iniciales para el estudio GO28141 se realizaron con una fecha de corte de datos del 9 de mayo de 2014. Se observó una mejoría significativa en la variable principal, SLP valorada por el Investigador, en los pacientes asignados al grupo de Cotellic más vemurafenib en comparación con el grupo de placebo más vemurafenib (HR 0,51 [0,39; 0,68]; valor p < 0,0001). La mediana estimada para la SLP valorada por el Investigador fue de 12,3 meses (95% IC 9,5, 13,4) para el grupo de Cotellic más vemurafenib versus 7,2 meses (95% IC 5,6, 7,5) para el de placebo más vemurafenib. La mediana de seguimiento de pacientes fue de 14,2 meses. La mediana estimada para la SLP por el CRI fue 11,3 meses para el grupo de Cotellic más vemurafenib versus 6,0 meses para el de placebo más vemurafenib (HR 0,60 [0,45; 0,79]; valor p = 0,0003). La tasa de respuesta objetiva (TRO) para el grupo de Cotellic más vemurafenib fue de 67,6% versus 44,8% para el de placebo más vemurafenib. La diferencia en TRO fue de 22,9% (valor p < 0,0001). El análisis de SG final para el Estudio GO28141 se realizó con fecha de corte de datos 28 de agosto de 2015. Se comprobó una mejoría significativa en SG en los pacientes asignados al grupo Cotellic más vemurafenib en comparación con el grupo de placebo más vemurafenib (Figura 1). Las SG estimadas a 1 año (75%) y a 2 años (48%) para el grupo Cotellic más vemurafenib fueron mayores que las pronosticadas para el grupo de placebo más vemurafenib (64% y 38% respectivamente).
El estado de salud global/la calidad de vida relacionada con la salud notificada por el paciente se midió mediante el Cuestionario de Calidad de Vida EORTC - Core 30 (QLQ-C30). Todos los ámbitos funcionales (cognitivo, emocional, social, rol y físico) y la mayor parte de los síntomas (pérdida de apetito, estreñimiento, insomnio, náuseas y vómitos, disnea, dolor, fatiga) fueron similares entre los dos grupos de tratamiento y no mostraron ningún cambio clínicamente significativo (todos los resultados fueron ≤ 10 puntos de cambio con respecto al inicio). Los pacientes en el grupo de Cotellic más vemurafenib reportaron empeoramiento significativo de la diarrea desde la línea de base sólo en el Ciclo 1 Día 15 y Ciclo 2 Día 15 según lo medido por EORTC QLQ-C30; pero no en puntos de tiempo posteriores. Estudio NO25395 (BRIM7): La eficacia de Cotellic se comprobó en el estudio Fase Ib NO25395, que se diseñó para evaluar la seguridad, tolerabilidad, farmacocinética y eficacia de Cotellic cuando se combina con vemurafenib para el tratamiento de pacientes con melanoma no resecable o metastásico con mutación BRAF V600 positiva (confirmada mediante el Test de Cobas® 4800 para la mutación BRAF V600). En este estudio se trataron 129 pacientes con Cotellic y vemurafenib: 63 no habían recibido anteriormente ningún tratamiento inhibidor del BRAF (BRAFi) y 66 habían presentado progresión con la terapia previa de vemurafenib. De los 63 pacientes sin tratamiento previo con BRAFi, 20 habían recibido una terapia sistémica anterior para melanoma avanzado, siendo la mayoría (el 80%) inmunoterapia. Los resultados obtenidos en la población sin tratamiento previo con BRAFi del estudio NO25395 concordaron en general con los del estudio GO28141. Estos pacientes (n = 63) alcanzaron una tasa de respuesta objetiva del 87%, incluidas las respuestas completas que se observaron en el 16% de los mismos. La mediana de duración de la respuesta fue de 14,3 meses. La mediana de sobrevida libre de progresión para los pacientes sin tratamiento previo con BRAFi fue de 13,8 meses, con una mediana de tiempo de seguimiento de 20,6 meses. Entre los pacientes que habían progresado con vemurafenib (n = 66), la tasa de respuesta objetiva fue del 15%, la mediana de duración de la respuesta de 6,8 meses y la mediana de la sobrevida libre de progresión de 2,8 meses, con una mediana de tiempo de seguimiento de 8,1 meses. En los pacientes sin tratamiento previo con inhibidor BRAF, la mediana de sobrevida global fue de 28,5 meses (IC del 95% 23,3-34,6). En los pacientes que habían progresado con la terapia del inhibidor BRAF, la mediana de supervivencia global fue de 8,4 meses (IC del 95% 6,7-11,1). Población pediátrica: La Agencia Europea de Medicamentos ha concedido al titular un aplazamiento para presentar los resultados de los ensayos realizados con Cotellic en uno o más grupos de población pediátrica en tumores sólidos malignos (véase Dosificación para consultar la información sobre el uso en la población pediátrica). Propiedades farmacocinéticas: Absorción: Después de la administración oral de 60 mg de cobimetinib en pacientes con cáncer, éste mostró una velocidad de absorción moderada, con una mediana de Tmáx de 2,4 horas. La media de la Cmáx en estado estacionario y del ABC0-24 fue de 273 ng/ml y 4.340 ng•h/ml, respectivamente. La mediana del cociente de acumulación en estado estacionario fue de aproximadamente 2,4 veces. Cobimetinib tiene una farmacocinética lineal en el rango de dosis de ~3,5 mg a 100 mg. La biodisponibilidad absoluta de cobimetinib fue del 45,9% (IC del 90%: 39,7%, 53,1%) en voluntarios sanos. Se realizó un estudio en seres humanos de balance de masa en individuos sanos, el cual demostró que cobimetinib es ampliamente metabolizado y excretado por las heces. La fracción absorbida fue de ~88%, indicando absorción elevada y metabolismo de primer paso. La farmacocinética de cobimetinib no se modifica cuando este fármaco se administra en estado posprandial (luego de una comida rica en grasas) en comparación con la ingesta en ayunas en voluntarios sanos. Dado que los alimentos no alteran la farmacocinética de cobimetinib, Cotellic puede administrarse con o sin ellos. Distribución: Cobimetinib se une en un 94,8% a las proteínas plasmáticas humanas in vitro. No se observó ninguna unión preferente a los hematíes en seres humanos (cociente sangre/plasma 0,93). El volumen de distribución fue de 1.050 litros en individuos sanos a los que se administró una dosis intravenosa de 2 mg. En pacientes con cáncer el volumen de distribución aparente fue de 806 litros, según lo indicado por el análisis de farmacocinética poblacional. Cobimetinib es un sustrato de P-gp in vitro. Se desconoce el transporte a través de la barrera hemato-encefálica. Biotransformación: La oxidación mediante CYP3A y la glucuronidación mediante UGT2B7 parecen ser las rutas metabólicas principales de cobimetinib. Cobimetinib es la fracción predominante en el plasma. No se observaron metabolitos oxidativos superiores al 10% de la radiactividad circulante total, ni metabolitos humanos específicos en el plasma. El fármaco inalterado en las heces y en la orina representó el 6,6% y el 1,6% de la dosis administrada, respectivamente, indicando que cobimetinib es principalmente metabolizado y tiene una eliminación renal mínima. In vitro los datos muestran que cobimetinib no es un inhibidor de OAT1, OAT3 u OCT2. Eliminación: Cobimetinib y sus metabolitos se caracterizaron en un estudio de balance de masa en sujetos sanos. Como promedio, se recuperó el 94% de la dosis en un plazo de 17 días. Cobimetinib en gran parte se metabolizó y se eliminó en forma de heces; no hubo un único metabolito predominante. Después de la administración intravenosa de una dosis de 2 mg de cobimetinib, el clearance plasmático promedio fue de 10,7 litros/hora. La mediana del clearance aparente luego de la administración oral de 60 mg en pacientes con cáncer fue de 13,8 litros/hora. El promedio de la vida media de eliminación después de la administración oral de cobimetinib fue de 43,6 horas (rango: 23,1 a 69,6 horas). En consecuencia, cobimetinib puede tardar hasta 2 semanas después de interrumpir el tratamiento en eliminarse completamente de la circulación sistémica. Farmacocinética en poblaciones especiales: Según lo indicado por un análisis de farmacocinética poblacional, el sexo, la raza, el origen étnico, el estado funcional ECOG basal y la insuficiencia renal leve y moderada no alteraron la farmacocinética de cobimetinib. Se identificaron la edad y el peso corporal basal como covariables estadísticamente significativas con respecto al clearance y el volumen de distribución de cobimetinib, respectivamente. Sin embargo, el análisis de sensibilidad sugiere que ninguna de estas dos covariables tuvo un impacto clínicamente significativo en la exposición en el estado estacionario. Sexo: El sexo carece de efectos sobre la exposición a cobimetinib, según lo indicado por un análisis de farmacocinética poblacional realizado en 210 mujeres y 277 hombres. Pacientes de edad avanzada: La edad no afecta la exposición a cobimetinib, de acuerdo con un análisis de farmacocinética poblacional llevado a cabo en 133 pacientes con 65 años de edad o mayores. Pacientes con insuficiencia renal: Basado en los datos preclínicos y el estudio de balance de masa en seres humanos, cobimetinib es principalmente metabolizado, con excreción renal mínima. No se ha conducido ningún estudio formal de farmacocinética en pacientes con insuficiencia renal. Un análisis de farmacocinética poblacional que utilizó datos de 151 pacientes con insuficiencia renal leve (clearance de creatinina [Clcr], 60 a menos de 90 ml/min), 48 con insuficiencia renal moderada (Clcr, 30 a menos de 60 ml/min) y 286 con función renal normal (ClCr, igual o superior a 90 ml/min), demostró que el Clcr no tuvo influencia significativa en la exposición a cobimetinib. La insuficiencia renal de leve a moderada no influye en la exposición a cobimetinib según lo indicado por el análisis farmacocinético poblacional. Dado que los datos existentes son limitados, no es posible determinar la necesidad de ajustes de dosis en pacientes con insuficiencia renal grave. Pacientes con insuficiencia hepática: Se evaluó la farmacocinética de cobimetinib en 6 sujetos con insuficiencia hepática leve (Child Pugh A), 6 con insuficiencia hepática moderada (Child Pugh B), 6 con insuficiencia hepática grave (Child Pugh C) y en 10 sujetos sanos. Las exposiciones sistémicas totales de cobimetinib después de una única dosis fueron similares en pacientes con insuficiencia hepática leve o moderada en comparación con los sujetos sanos, mientras que los que padecían insuficiencia hepática grave tuvieron exposiciones totales a cobimetinib inferiores (proporción de media geométrica AUC0-inf 0,69 en comparación con sujetos sanos) que no se consideran clínicamente significativas. Las exposiciones de cobimetinib no ligado fueron semejantes entre sujetos con insuficiencia hepática moderada y leve en comparación con la de los que tenían función hepática normal, mientras que aquéllos con insuficiencia hepática grave tuvieron aproximadamente exposiciones dos veces más altas (ver Dosificación). Por lo tanto, no se recomienda un ajuste de la dosis cuando se administra Cotellic a pacientes con insuficiencia hepática (ver Dosificación y Precauciones). Población pediátrica: No se ha realizado ningún estudio para investigar la farmacocinética de cobimetinib en esta población. Datos preclínicos sobre seguridad: No se han llevado a cabo estudios de carcinogenicidad con cobimetinib. Los ensayos de genotoxicidad estándares realizados con cobimetinib dieron resultado negativo. No se ha efectuado ningún estudio de fertilidad específico en animales con cobimetinib. En los ensayos de toxicología en dosis repetidas, se observaron cambios degenerativos en los tejidos reproductores, incluyendo aumento de la apoptosis/necrosis de los cuerpos lúteos y las vesículas seminales, células epiteliales epididimarias y vaginales en ratas, y células epiteliales epididimarias en perros. Se desconoce su importancia clínica. Cuando se administró a ratas preñadas, cobimetinib causó mortalidad embrionaria y malformaciones fetales de los grandes vasos y del cráneo en exposiciones sistémicas similares a la exposición en seres humanos con la dosis recomendada. No se ha evaluado la seguridad cardiovascular de cobimetinib en combinación con vemurafenib in vivo. In vitro, cobimetinib produjo una inhibición moderada del canal del ion hERG (IC50 = 0,5 mM [266 ng/ml]), lo cual es aproximadamente 18 veces superior a las concentraciones plasmáticas máximas (Cmáx) alcanzadas con la dosis de 60 mg (Cmáx del fármaco no ligado = 14 ng/ml [0,03 mM]). Los estudios de toxicidad en ratas y perros identificaron, en general, cambios degenerativos reversibles en la médula ósea, el tracto gastrointestinal, la piel, el timo, la glándula suprarrenal, el hígado, el bazo, los ganglios linfáticos, los riñones, el corazón, los ovarios y la vagina, en exposiciones plasmáticas inferiores a los niveles clínicos eficaces. Los efectos tóxicos que limitan la dosis incluyen ulceraciones cutáneas, exudados superficiales y acantosis en ratas e inflamación activa crónica y degeneración del esófago asociada con grados variables de gastroenteropatía en perros. En un estudio de toxicidad de dosis repetidas en ratas jóvenes, las exposiciones sistémicas a cobimetinib fueron de 2 a 11 veces superiores a los 10 días después de nacer que a los 38 días, cuando las exposiciones fueron similares a las de las ratas adultas. En las ratas jóvenes, la administración de cobimetinib dio lugar a cambios semejantes a los vistos en los estudios de toxicidad pivotales en ratas adultas, incluyendo modificaciones degenerativas reversibles en el timo y el hígado, disminución del peso de la tiroides/paratiroides y del bazo, aumento del fósforo, la bilirrubina y la masa sanguínea de glóbulos rojos, y descenso de los triglicéridos. En animales jóvenes se produjo una mortalidad con una dosis (3 mg/kg) que no generó ningún deceso en animales adultos. Los estudios preclínicos no muestran otros riesgos especiales para los seres humanos según los estudios convencionales de farmacología de seguridad y genotoxicidad.
Indicaciones.
Cotellic está indicado en combinación con vemurafenib para el tratamiento de pacientes adultos con melanoma no resecable o metastásico con una mutación BRAF V600 (véanse Precauciones y Farmacología, Propiedades farmacodinámicas).
Dosificación.
General: El tratamiento con Cotellic en combinación con vemurafenib debe ser iniciado y supervisado exclusivamente por un profesional de la salud con experiencia en el tratamiento de pacientes con cáncer. Antes de comenzar este tratamiento, los pacientes deben tener un diagnóstico de mutación BRAF V600 positiva en el tumor, confirmado con una prueba validada (véanse Precauciones y Farmacología, Propiedades farmacodinámicas). Posología: La dosis recomendada de Cotellic es de 60 mg (3 comprimidos de 20 mg) una vez por día. Cotellic se toma en ciclos de 28 días. Cada dosis consiste en tres comprimidos de 20 mg (60 mg) y se deben tomar una vez por día durante 21 días consecutivos (días 1 a 21: período de tratamiento); seguidos de un descanso de 7 días (días 22 a 28: pausa del tratamiento). Cada ciclo siguiente del tratamiento con Cotellic se debería iniciar después de que haya terminado el descanso de 7 días sin tratamiento. Para información sobre la posología de vemurafenib, consulte el Prospecto Información para Profesionales. Duración del tratamiento: El tratamiento con Cotellic deberá continuarse hasta que deje de aportar un efecto beneficioso al paciente o se produzca una toxicidad inaceptable (véase la Tabla 2). Dosis omitidas o retrasadas: Si se omite la toma de una dosis, puede tomarse más tarde hasta un máximo de 12 horas previas a la siguiente dosis, para mantener la pauta de administración una vez por día. Vómitos: En el caso de que se produzcan vómitos después de la administración de Cotellic, el paciente no deberá tomar una dosis adicional ese día, y continuará el tratamiento al día siguiente según lo prescripto. Modificaciones generales de la dosis: La decisión de reducir o no la dosis de uno o ambos tratamientos deberá basarse en la evaluación realizada por el prescriptor de la seguridad o la tolerabilidad individual del paciente. La modificación de la dosis de Cotellic es independiente de la modificación de la dosis de vemurafenib. Si se omiten dosis por motivos de toxicidad, estas dosis no deberán sustituirse. Una vez reducida la dosis, no deberá aumentarse con posterioridad. A continuación la Tabla 2 proporciona una recomendación general para la modificación de la dosis de Cotellic.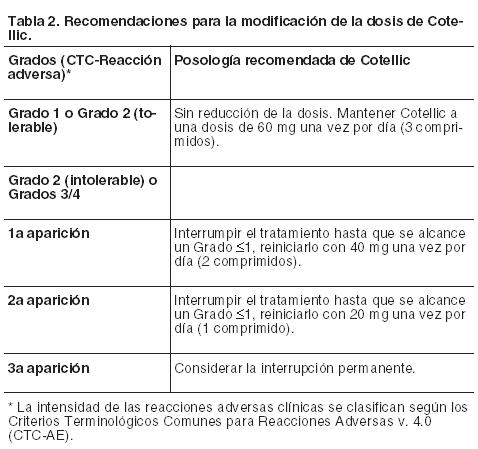
Recomendaciones sobre la modificación de la dosis para reacciones adversas especificadas: Hemorragia: Eventos Grado 4 o hemorragia cerebral (todos los grados): Interrumpir el tratamiento con Cotellic. Discontinuar permanentemente Cotellic por eventos de hemorragia atribuidos al mismo. Eventos Grado 3: Interrumpir el tratamiento con Cotellic. No hay datos sobre la eficacia de la modificación de la dosis de Cotellic por eventos de hemorragia. Debe aplicarse el criterio clínico al considerar reiniciar el tratamiento con Cotellic. Si está clínicamente indicado, la administración de vemurafenib puede continuarse cuando se interrumpe el tratamiento con Cotellic. Disfunción ventricular izquierda: Deberá considerarse la discontinuación del tratamiento con Cotellic en el caso de que los síntomas cardíacos se atribuyan a este medicamento y estos no mejoran después de la interrupción temporal del fármaco.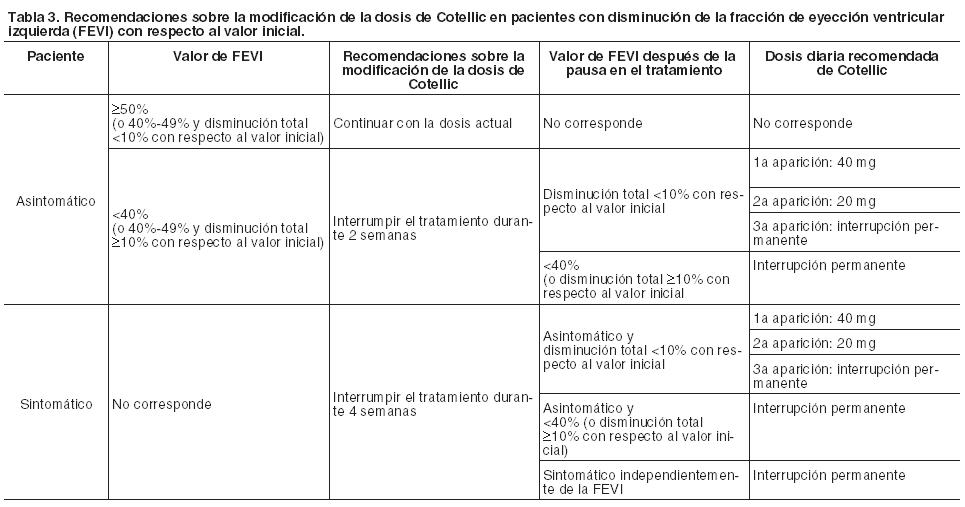
El tratamiento con vemurafenib puede continuarse cuando se modifique el tratamiento con Cotellic, si está clínicamente indicado. Recomendaciones sobre la modificación de la dosis de Cotellic cuando se utiliza con vemurafenib. Alteraciones analíticas hepáticas: En el caso de alteraciones analíticas hepáticas de Grados 1 y 2, se debe continuar el tratamiento con Cotellic y vemurafenib según la dosis prescripta. Grado 3: Se debe continuar el tratamiento con Cotellic según la dosis prescripta. Se puede reducir la dosis de vemurafenib cuando resulte clínicamente adecuado (consultar el Prospecto Información para Profesionales de vemurafenib). Grado 4: Se debe interrumpir el tratamiento con Cotellic y con vemurafenib. Si las alteraciones analíticas hepáticas mejoran hasta un Grado ≤ 1 dentro de las 4 semanas, se debe reiniciar el tratamiento con Cotellic con una dosis reducida de 20 mg y con vemurafenib a una dosis clínicamente adecuada, según su Prospecto Información para Profesionales. Se debe suspender el tratamiento con Cotellic y con vemurafenib si las alteraciones analíticas hepáticas no se resuelven hasta un Grado ≤ 1 dentro de las 4 semanas o si reaparecen las alteraciones analíticas hepáticas de Grado 4 después de la mejoría inicial. Rabdomiólisis y elevaciones de creatinina fosfoquinasa (CPK): Rabdomiólisis o elevaciones de CPK sintomáticas: Interrupción del tratamiento con Cotellic. Si la gravedad mejora por lo menos un grado dentro de las 4 semanas, reinicie Cotellic a una dosis reducida de 20 mg, de estar clínicamente indicado. La dosificación de vemurafenib se puede continuar si se modifica el tratamiento con Cotellic, de estar clínicamente indicado. Si la rabdomiólisis o las elevaciones de CPK sintomáticas no mejoran dentro de las 4 semanas, suspender en forma permanente el tratamiento con Cotellic. Elevaciones de CPK asintomáticas: Grado ≤ 3: No se debe modificar ni suspender la dosis de Cotellic para controlar elevaciones de CPK asintomáticas Grado ≤ 3 (véase Reacciones adversas). Grado 4: Interrupción del tratamiento con Cotellic. Si mejora a Grado ≤ 3 dentro de las 4 semanas, reinicie Cotellic a una dosis reducida de 20 mg, si está clínicamente indicado. La dosificación de vemurafenib se puede continuar si se modifica el tratamiento con Cotellic, si está clínicamente indicado. Si los niveles elevados de CPK no mejoran al Grado ≤ 3 en las 4 semanas después de la interrupción de dosis, suspender en forma permanente el tratamiento con Cotellic. Fotosensibilidad: Se debe controlar la fotosensibilidad de Grado ≤ 2 (tolerable) con cuidados complementarios. Fotosensibilidad de Grado 2 (intolerable) o de Grado ≥ 3: Se interrumpirá el tratamiento con Cotellic y vemurafenib hasta que se resuelva a Grado ≤ 1. El tratamiento puede reiniciarse sin modificar la dosis de Cotellic y deberá reducirse la dosis de vemurafenib según resulte clínicamente adecuado (consultar el Prospecto Información para Profesionales de vemurafenib). Erupción: Se pueden producir casos de erupciones debidas tanto al tratamiento con Cotellic como a vemurafenib. Se puede interrumpir y/o reducir temporalmente la dosis de Cotellic y/o vemurafenib cuando esté clínicamente indicado. Además, en caso de: Una erupción de Grado ≤ 2 (tolerable): deberá tratarse con cuidados complementarios. Se puede continuar la administración de la dosis de Cotellic sin modificaciones. Una erupción acneiforme de Grado 2 (intolerable) o de Grado ≥ 3: Se deben seguir las recomendaciones generales de las modificaciones de dosis de Cotellic que figuran en la Tabla 2. Se puede continuar la administración de la dosis de vemurafenib cuando se modifique el tratamiento con Cotellic, si está clínicamente indicado. Erupción no acneiforme o maculopapular de Grado 2 (intolerable) o Grado ≥ 3: Se puede continuar sin modificaciones la administración de la dosis de Cotellic si está clínicamente indicado. La dosis de vemurafenib se puede interrumpir y/o reducir temporalmente, para mayor información consultar su Prospecto Información para Profesionales. Prolongación del intervalo QT: Si durante el tratamiento el intervalo QTc es superior a 500 ms, por favor consulte el Prospecto Información para Profesionales (Dosificación) para modificar la dosis de vemurafenib. No se requiere modificar la dosis de Cotellic cuando se toma en combinación con vemurafenib. Poblaciones especiales: Pacientes de edad avanzada: No es necesario ningún ajuste de la dosis de Cotellic en los pacientes con 65 años de edad o mayores. Pacientes pediátricos: No se ha establecido la seguridad y eficacia de Cotellic en niños y adolescentes menores de 18 años de edad. No se dispone de datos. Pacientes con insuficiencia renal: No se recomienda ningún ajuste de la dosis en pacientes con insuficiencia renal leve o moderada, según los resultados obtenidos en un análisis de farmacocinética poblacional (véase Farmacología, Propiedades farmacocinéticas). Se dispone de pocos datos sobre Cotellic en pacientes con insuficiencia renal grave, por lo que no se puede descartar algún efecto. Cotellic se debe utilizar con precaución en pacientes con esta patología. Pacientes con insuficiencia hepática: No se aconseja ningún ajuste de dosis en pacientes con insuficiencia hepática. Los pacientes con insuficiencia hepática grave podrían presentar concentraciones plasmáticas elevadas de cobimetinib no ligado en comparación con las de aquellos con una función hepática normal (véase Farmacología; Propiedades farmacocinéticas). Se pueden producir alteraciones analíticas hepáticas cuando se usa Cotellic, por lo que se debe tener precaución en pacientes con insuficiencia hepática de cualquier grado (véase Precauciones). Pacientes no caucásicos: No se ha establecido la seguridad y eficacia de Cotellic en pacientes no caucásicos. Formas de administración: Cotellic es para uso por vía oral. Los comprimidos se deben ingerir enteros con agua. Se pueden tomar con o sin alimentos.
Contraindicaciones.
Cotellic está contraindicado en pacientes con hipersensibilidad conocida a cobimetinib o a cualquiera de sus excipientes.
Reacciones adversas.
Resumen del perfil de seguridad: En el estudio GO28141 se evaluó en 247 pacientes con melanoma avanzado con mutación BRAF V600 la seguridad de Cotellic en combinación con vemurafenib. La mediana de tiempo hasta la aparición del primer evento adverso de Grado ≥ 3 fue de 0,6 meses en el grupo tratado con Cotellic más vemurafenib frente a los 0,8 meses del grupo tratado con placebo más vemurafenib. En el estudio NO25395 también se evaluó la seguridad de Cotellic en combinación con vemurafenib en 129 pacientes con melanoma avanzado con mutación BRAF V600. El perfil de seguridad de este estudio concordó con el observado en el estudio GO28141. En el estudio GO28141, las reacciones adversas más comunes (20%) que se observaron con una frecuencia mayor en el grupo de Cotellic más vemurafenib fueron diarrea, rash, náuseas, fiebre, reacción de fotosensibilidad, aumento de alanina aminotransferasa y de aspartato aminotransferasa, incremento en sangre de creatinina fosfoquinasa y vómitos. Las reacciones adversas más comunes (20%) que se verificaron con una frecuencia mayor en el grupo de placebo más vemurafenib fueron artralgia, alopecia e hiperqueratosis. La fatiga se registró en ambos grupos de manera similar. Para una descripción completa de todas las reacciones adversas asociadas con el tratamiento con vemurafenib, consultar su Prospecto Información para Profesionales. Tabla de reacciones adversas: Las reacciones adversas (RAM) se basan en los resultados de un estudio de Fase III (GO28141), multicéntrico, aleatorizado, doble-ciego, controlado con placebo que evaluó la seguridad y eficacia de Cotellic en combinación con vemurafenib en comparación con vemurafenib en monoterapia en pacientes con mutación BRAF V600 positiva que no habían sido previamente tratados y que padecían melanoma no resecable localmente avanzado (estadio IIIc) o melanoma metastásico (estadio IV). Las frecuencias de las reacciones adversas (RAM) se basan en los análisis de seguridad de los pacientes tratados con cobimetinib mas vemurafenib con una mediana de seguimiento de 11,2 meses (fecha de corte de datos 19 de septiembre de 2014). Las reacciones adversas (RAM) que fueron notificadas en los pacientes con melanoma se han enumerado bajo la clasificación por órganos y sistemas de MedDRA, frecuencia y grado de gravedad. Se han utilizado las siguientes categorías para clasificar la frecuencia de aparición: muy frecuentes (≥1/10), frecuentes (≥1/100 a < 1/10), poco frecuentes (≥1/1.000 a < 1/100), raras (≥1/10.000 a < 1/1.000) y muy raras ( < 1/10.000). La Tabla 4 resume las reacciones adversas relacionadas con el uso de Cotellic. Dentro de cada grupo de frecuencia, se presentan las reacciones adversas en orden decreciente de gravedad y se informaron de acuerdo con NCI-CTCAE versión 4.0 (Criterios Comunes de Toxicidad) para evaluar la toxicidad en el estudio GO28141.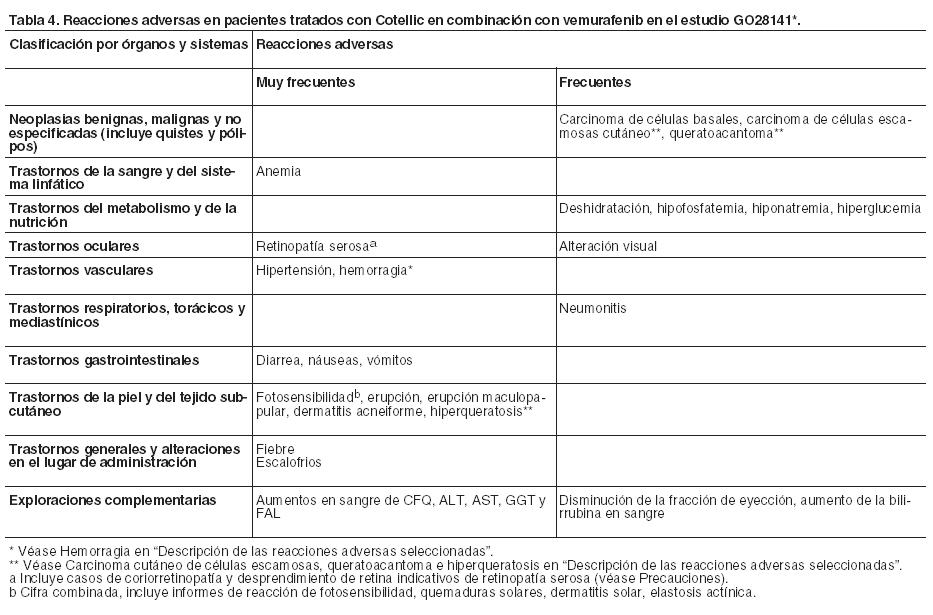
Información adicional sobre reacciones adversas seleccionadas: Hemorragia: Los episodios de sangrado se han registrado con mayor frecuencia en el grupo de Cotellic más vemurafenib que en el de placebo más vemurafenib (todos los tipos y Grados: 13% frente a 7%). En el grupo de Cotellic más vemurafenib se observaron frecuencias mayores de hemorragia cerebral (1% frente a 0%), hemorragia del tracto gastrointestinal (4% frente a 1%), hemorragia en el sistema reproductor (2% frente a ≤1%) y hematuria (3% frente a 1%). La mayoría de los eventos no fueron graves y de Grados 1 ó 2 (12% de los pacientes en el grupo de Cotellic más vemurafenib en comparación con el 7% de los de placebo más vemurafenib). Un 1% de los pacientes de cada brazo sufrió casos de Grados 3-4 (véase Precauciones). La mediana de tiempo hasta el primer acontecimiento fue de 4,4 meses (rango de 0,0 a 12,7 meses) en el grupo tratado con Cotellic más vemurafenib. La mayoría de los eventos se resolvieron o fueron resueltos sin cambios en la dosis Cotellic. Fotosensibilidad: Se observó fotosensibilidad con mayor frecuencia en el grupo de Cotellic más vemurafenib que en el de placebo más vemurafenib (47% frente a 35%). La mayoría de los eventos fueron de Grados 1 ó 2, mientras que los de Grado ≥ 3 ocurrieron en el 4% de los pacientes en el grupo de Cotellic más vemurafenib en comparación con el 0% en el de placebo más vemurafenib. No se apreció ninguna tendencia en lo relativo al tiempo transcurrido hasta el inicio de los episodios de Grado ≥ 3. Los eventos adversos de fotosensibilidad de Grado ≥ 3 en el grupo de Cotellic más vemurafenib fueron tratados con medicación tópica primaria junto con la interrupción de las dosis tanto de Cotellic como de vemurafenib (véase Dosificación). No se verificó indicio alguno de fototoxicidad con el empleo del cobimetinib en monoterapia. Carcinoma de células escamosas cutáneo, queratoacantoma e hiperqueratosis: Se han informado casos de carcinoma de células escamosas cutáneo con menor frecuencia en el grupo de Cotellic más vemurafenib en comparación con el grupo de placebo más vemurafenib (todos los Grados: 3% frente a 13%). Se informaron casos de queratoacantoma con una frecuencia inferior en el grupo de Cotellic más vemurafenib en comparación con el de placebo más vemurafenib (todos los Grados: 2% frente a 9%). Los casos de hiperqueratosis se registraron con una frecuencia inferior en el grupo de Cotellic más vemurafenib en comparación con el de placebo más vemurafenib (todos los Grados: 11% frente a 30%). Retinopatía serosa: Se han comunicado casos de retinopatía serosa en pacientes tratados con Cotellic (véase Precauciones). Para pacientes que notifican trastornos visuales nuevos o empeoramiento de los mismos, se recomienda un examen oftalmológico. La retinopatía serosa se puede controlar mediante la interrupción del tratamiento, la reducción de la dosis o la suspensión del tratamiento (véase Dosificación, Tabla 2). Disfunción ventricular izquierda: Se han notificado casos de disminución de la FEVI con respecto al inicio en pacientes tratados con Cotellic (véase Precauciones). Se debe evaluar la FEVI antes de iniciar la administración del fármaco para establecer los valores de referencia, y posteriormente, después del primer mes de tratamiento y, como mínimo, cada 3 meses o cuando esté clínicamente indicado hasta la suspensión del mismo. La disminución de la FEVI con respecto al inicio se puede controlar mediante la interrupción del tratamiento, la reducción de la dosis o la suspensión del tratamiento (véase Dosificación). Alteraciones según las pruebas de laboratorio: Alteraciones analíticas hepáticas: Se han observado alteraciones analíticas hepáticas, específicamente ALT, AST y FAL, en pacientes tratados con Cotellic en combinación con vemurafenib (véase Precauciones). Se deben monitorizar las pruebas analíticas hepáticas antes de iniciar el tratamiento en combinación y cada mes durante el tratamiento, o con mayor frecuencia si está clínicamente indicado (véase Dosificación). Aumento de la creatinina fosfoquinasa en sangre: Se registraron aumentos asintomáticos de los niveles de CPK en sangre con mayor frecuencia en el grupo tratado con Cotellic más vemurafenib respecto del que recibió placebo más vemurafenib en el estudio GO28141 (véase Dosificación). Se observó un caso de rabdomiólisis en cada grupo de tratamiento en este estudio con los consecuentes aumentos de CPK en sangre. La Tabla 5 muestra la frecuencia de las alteraciones analíticas hepáticas medidas y el aumento de creatinina fosfoquinasa para todos los Grados y los Grados 3-4.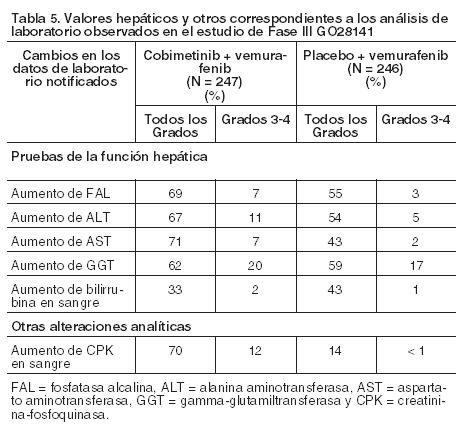
Experiencia poscomercialización: Reacciones adversas notificadas en la experiencia poscomercialización:
Poblaciones especiales: Pacientes de edad avanzada: En el estudio Fase III con Cotellic en combinación con vemurafenib en pacientes con melanoma no resecable o metastásico (n = 257), 183 pacientes (74%) tenían 65 años de edad, 44 (18%) entre 65 y 74 años, 16 (6%) entre 75 y 84 años, y 4 (2%) 85 años o mayores. La proporción de pacientes que sufrieron reacciones adversas (RAM) fue similar en pacientes de 65 años que en aquéllos de 65 años o mayores. Los pacientes de 65 años o mayores fueron más propensos que los de 65 años a sufrir reacciones adversas graves y eventos adversos que produjeron la suspensión del tratamiento con cobimetinib. Pacientes con insuficiencia renal: No se ha llevado a cabo ningún ensayo farmacocinético en sujetos con insuficiencia renal. No se recomienda ajustar la dosis para la insuficiencia renal de leve a moderada en base a los resultados del análisis farmacocinético poblacional. Se dispone de pocos datos sobre Cotellic en pacientes con insuficiencia renal grave. Cotellic se debe usar con precaución en pacientes con esta patología. Pacientes con insuficiencia hepática: No se recomienda ningún ajuste de dosis en pacientes con insuficiencia hepática (véase Farmacología; Propiedades farmacocinéticas). Comunicación de reportes de reacciones adversas: Es importante comunicar las presuntas reacciones adversas después de la autorización del medicamento. Esto permite la monitorización continua de la relación riesgo/beneficio. Se solicita a los profesionales de la salud informar de cualquier sospecha de eventos adversos asociados con el uso de Cotellic® al Área de Farmacovigilancia de Roche al siguiente teléfono 0800-77-ROCHE (76243). En forma alternativa, esta información puede ser reportada ante ANMAT. Ante cualquier inconveniente con el producto, el paciente puede llenar la ficha que está en la Página Web de la ANMAT: http://www.anmat.gov.ar/farmacovigilancia/Notificar.asp o llamar a ANMAT responde al 0800-333-1234.
Precauciones.
Antes de tomar Cotellic en combinación con vemurafenib, se debe haber confirmado por una prueba validada que los pacientes tienen un tumor con una mutación BRAF V600 positiva. Cotellic en combinación con vemurafenib en pacientes que han progresado con un inhibidor de BRAF. Existen pocos datos