HARVONI®
GADOR
Grupo farmacoterapéutico: antiviral de acción directa, código ATC: J05AP51.
Composición.
Cada comprimido recubierto contiene: Ledipasvir 90 mg, Sofosbuvir 400 mg. Excipientes: Copovidona, Lactosa monohidrato, Celulosa microcristalina, Croscarmelosa sódica, Dióxido de silicio coloidal, Estearato de magnesio, Alcohol polivinilico parcialmente hidrolizado*, Dióxido de titanio*, Polietilenglicol 3350*, Talco*, Colorante FD&C Amarillo N°6* c.s. * Se refiere a los componentes del Opadry II anaranjado 85F13912. Excipientes con efecto conocido: HARVONI® contiene lactosa monohidrato y Colorante FD&C amarillo N°6.
Farmacología.
Descripción: Los comprimidos de HARVONI® son comprimidos de asociación en dosis fijas, que contienen ledipasvir y sofosbuvir. Ledipasvir es un inhibidor del VHC que actúa sobre la proteína NS5A de dicho virus, que es esencial tanto para la replicación del ARN, como para el ensamblaje de los viriones del VHC. Sofosbuvir es un inhibidor pangenotípico de la polimerasa de ARN dependiente del ARN NS5B del VHC, que es esencial para la replicación viral. Sofosbuvir es un profármaco nucleotídico que sufre metabolismo intracelular para formar el trifosfato análogo de la uridina farmacológicamente activo (GS-461203), que puede ser incorporado al ARN del VHC por la polimerasa NS5B y actúa como terminador de cadena. Comprimido recubierto con película de color anaranjado y forma de rombo, y con "GSI" grabado por un lado y "7985" por el otro. Los comprimidos de HARVONI® se administran por vía oral. Cada comprimido recubierto contiene 90 mg de ledipasvir y 400 mg de sofosbuvir como principios activos. Los comprimidos también incluyen los siguientes excipientes: copovidona, lactosa monohidrato, celulosa microcristalina, croscarmelosa sódica, dióxido de silicio coloidal y estearato de magnesio. Los comprimidos están recubiertos con Alcohol polivinílico, Dióxido de titanio, Polietilenglicol 3350, Talco, y Colorante FD&C amarillo N°6. Farmacología clínica: Mecanismo de acción: Ledipasvir es un inhibidor del VHC que actúa sobre la proteína NS5A de dicho virus, que es esencial tanto para la replicación del ARN, como para el ensamblaje de los viriones del VHC. La confirmación bioquímica de la inhibición de NS5A por parte de ledipasvir no es posible en la actualidad, ya que NS5A carece de función enzimática. Los ensayos in vitro de selección de resistencias y resistencia cruzada indican que el efecto de ledipasvir sobre la NS5A es su modo de acción. Sofosbuvir es un inhibidor pangenotípico de la polimerasa de ARN dependiente del ARN NS5B del VHC, que es esencial para la replicación viral. Sofosbuvir es un profármaco nucleotídico que sufre metabolismo intracelular para formar el trifosfato análogo de la uridina farmacológicamente activo (GS-461203), que puede ser incorporado al ARN del VHC por la polimerasa NS5B y actúa como terminador de cadena. GS-461203 (el metabolito activo de sofosbuvir) no es un inhibidor de las polimerasas de ADN y ARN humanas ni un inhibidor de la polimerasa de ARN mitocondrial. Farmacocinética: Absorción: Tras la administración oral de ledipasvir/sofosbuvir a pacientes infectados por el VHC, la mediana de las concentraciones plasmáticas máximas de ledipasvir se observó a las 4,0 horas después de la administración. Sofosbuvir se absorbió rápidamente y la mediana de las concentraciones plasmáticas máximas se observó aproximadamente 1 hora después de la administración. La mediana de la concentración plasmática máxima de GS-331007 se observó a las 4 horas después de la administración. Según los análisis farmacocinéticos poblacionales en pacientes infectados por el VHC, las medias geométricas del AUC0-24 de ledipasvir (n = 2.113), sofosbuvir (n = 1.542) y GS-331007 (n = 2.113) en situación de equilibrio fueron de 7.290, 1.320 y 12.000 ng•h/ml, respectivamente. Las Cmáx de ledipasvir, sofosbuvir y GS-331007 en situación de equilibrio fueron de 323, 618 y 707 ng/ml, respectivamente. El AUC0-24 y la Cmáx de sofosbuvir y GS-331007 fueron similares en los sujetos adultos sanos y en los pacientes con infección por el VHC. En comparación con los sujetos sanos (n = 191), el AUC0-24 y la Cmáx de ledipasvir fueron un 24 % y un 32 % más bajas, respectivamente, en los pacientes infectados por el VHC. El AUC de ledipasvir es proporcional a la dosis a lo largo del intervalo posológico comprendido entre 3 y 100 mg. Las AUC de sofosbuvir y GS-331007 son casi proporcionales a la dosis a lo largo del intervalo posológico comprendido entre 200 mg y 400 mg. Efectos de los alimentos: En comparación con las condiciones de ayuno, la administración de una dosis única de ledipasvir/sofosbuvir con una comida de contenido graso moderado o alto aumentó el AUC0-inf de sofosbuvir aproximadamente en 2 veces, pero no afectó significativamente a la Cmáx de sofosbuvir. Las exposiciones a GS-331007 y ledipasvir no se alteraron como consecuencia de ninguno de los tipos de comida. HARVONI® se puede administrar sin tener en cuenta la ingesta de alimentos. Distribución: Ledipasvir se une a las proteínas plasmáticas humanas en > 99,8 %. Tras una dosis única de 90 mg de [14C]-ledipasvir en sujetos sanos, el cociente sangre/plasma de radioactividad [14C] osciló entre 0,51 y 0,66. Sofosbuvir se une a las proteínas plasmáticas humanas en aproximadamente un 61-65 % y la unión es independiente de la concentración del fármaco a lo largo del intervalo comprendido entre 1 mg/ml y 20 mg/ml. La unión de GS-331007 a proteínas fue mínima en el plasma humano. Tras una dosis única de 400 mg de [14C]-sofosbuvir en sujetos sanos, el cociente sangre/plasma de radioactividad [14C] fue de aproximadamente 0,7. Biotransformación: In vitro, no se observó un metabolismo detectable de ledipasvir por parte de las enzimas CYP1A2, CYP2C8, CYP2C9, CYP2C19, CYP2D6 y CYP3A4 humanas. Se han observado datos indicativos de un metabolismo oxidativo lento a través de un mecanismo desconocido. Tras una dosis única de 90 mg de [14C]-ledipasvir, la exposición sistémica se debió casi exclusivamente al fármaco parental ( > 98 %). El ledipasvir inalterado también es la principal forma presente en las heces. Sofosbuvir se metaboliza ampliamente en el hígado para formar el trifosfato análogo del nucleósido farmacológicamente activo GS-461203. No se observa el metabolito activo. La vía de activación metabólica engloba una hidrólisis secuencial del resto de carboxiléster catalizado por la catepsina A humana o la carboxilesterasa 1 y una escisión del fosforamidato por parte de la proteína fijadora de nucleótidos de la tríada de la histidina 1 seguida de una fosforilación a través de la vía de biosíntesis de nucleótidos de pirimidina. La desfosforilación da lugar a la formación del metabolito nucleosídico GS-331007, que no se puede refosforilar eficazmente y carece de actividad contra el VHC in vitro. En ledipasvir/sofosbuvir, GS-331007 representa aproximadamente el 85 % de la exposición sistémica total. Eliminación: Tras una dosis única por vía oral de 90 mg de [14C]-ledipasvir, la recuperación total media de la radioactividad [14C] en las heces y en la orina fue del 87 %, con la mayoría de la dosis radioactiva recuperada en las heces (el 86 %). El ledipasvir inalterado excretado con las heces representó una media del 70 % de la dosis administrada y el metabolito oxidativo M19 representó un 2,2 % de la dosis. Estos datos sugieren que la excreción biliar de ledipasvir inalterado es una vía de eliminación principal y que la excreción renal es una vía de escasa importancia (aproximadamente un 1 %). La mediana de la semivida terminal de ledipasvir en voluntarios sanos tras la administración de ledipasvir/sofosbuvir en ayunas fue de 47 horas. Tras una dosis única por vía oral de 400 mg de [14C]-sofosbuvir, la recuperación total media de la dosis fue superior al 92 %, con una recuperación de aproximadamente el 80 %, el 14 % y el 2,5 % en orina, heces y aire espirado, respectivamente. La mayor parte de la dosis de sofosbuvir recuperada en la orina fue GS-331007 (78 %), mientras que el 3,5 % se recuperó en forma de sofosbuvir. Estos datos indican que la depuración renal es la principal vía de eliminación de GS-331007, con una gran parte secretada activamente. La mediana de la semivida terminal de sofosbuvir y de GS-331007 tras la administración de ledipasvir/sofosbuvir fue de 0,5 y 27 horas, respectivamente. Ni ledipasvir ni sofosbuvir son sustratos de los transportadores de captación hepática, transportador de cationes orgánicos (TCO) 1, polipéptido transportador de aniones orgánicos (PTAO) 1B1 o PTAO1B3. GS-331007 no es un sustrato de los transportadores renales, incluidos el transportador de aniones orgánicos (TAO) 1, TAO3 o TCO2. Potencial de ledipasvir/sofosbuvir in vitro para afectar a otros medicamentos: En las concentraciones alcanzadas en la práctica clínica, ledipasvir no es un inhibidor de los transportadores hepáticos, incluidos el PTAO1B1 o PTAO1B3, BSEP, OCT1, OCT2, TAO1, TAO3, transportador de extrusión de múltiples fármacos y compuestos tóxicos (EMFCT) 1, proteína de resistencia a múltiples fármacos (PRMF) 2 o PRMF4. Sofosbuvir y GS-331007 no son inhibidores de los transportadores de fármacos glucoproteína P y la proteína de resistencia del cáncer de mama (PRCM), PRMF2, BESB, PTAO1B1, PTAO1B3 y TCO1 y GS-331007 no es un inhibidor de TAO1, TCO2 y EMFCT1. Sofosbuvir y GS-331007 no son inhibidores ni inductores de las enzimas del CYP ni de la uridina difosfato glucuronosiltransferasa (UGT) 1A1. Farmacocinética en poblaciones especiales: Raza y sexo: No se han identificado diferencias farmacocinéticas clínicamente relevantes debidas a la raza para ledipasvir, sofosbuvir o GS-331007. No se han identificado diferencias farmacocinéticas clínicamente relevantes debidas al sexo para sofosbuvir o GS-331007. El AUC y la Cmáx de ledipasvir fueron un 77 % y un 58% más altas, respectivamente, en las mujeres que en los hombres; no obstante, la relación entre sexo y exposición a ledipasvir no se consideró clínicamente relevante. Personas de edad avanzada: Los análisis farmacocinéticos poblacionales en pacientes infectados por el VHC mostraron que dentro del intervalo de edad estudiado (18 a 80 años), la edad no tuvo un efecto clínicamente relevante sobre la exposición a ledipasvir, sofosbuvir o a GS-331007. Los ensayos clínicos con ledipasvir/sofosbuvir incluyeron a 235 pacientes de edad igual o superior a 65 años (el 8,6% del número total de pacientes). Insuficiencia renal: En la Tabla 1 se muestra un resumen del efecto de diferentes grados de insuficiencia renal (IR) sobre las exposiciones a los componentes de HARVONI® comparado con sujetos con función renal normal, como se describe en el texto a continuación. (Ver Tabla 1).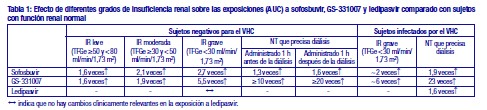
La farmacocinética de ledipasvir se estudió con una dosis única de 90 mg del medicamento en pacientes negativos para el VHC con insuficiencia renal grave (TFGe < 30 ml/min por Cockcroft-Gault, mediana [intervalo] del ClCr 22 [17-29] ml/min). La farmacocinética de sofosbuvir se estudió en pacientes negativos para el VHC con insuficiencia renal leve (TFGe ≥50 y < 80 ml/min/1,73m²), moderada (TFGe ≥30 y < 50 ml/min/1,73m²) y grave (TFGe < 30 ml/min/1,73 m²) y en pacientes con nefropatía terminal (NT) que precisan hemodiálisis tras una dosis única de 400 mg de sofosbuvir. En comparación con los pacientes con función renal normal (TFGe > 80 ml/min/1,73 m²), GS-331007 se elimina eficazmente mediante hemodiálisis, con un coeficiente de extracción de aproximadamente el 53 %. Tras una dosis única de 400 mg de sofosbuvir, una hemodiálisis de 4 horas eliminó el 18 % de la dosis de sofosbuvir administrada. En pacientes infectados con VHC con insuficiencia renal grave tratados con ledipasvir / sofosbuvir durante 12 semanas (n = 18), la farmacocinética de ledipasvir, sofosbuvir y GS-331007 fue consistente con la observada en pacientes negativos para VHC con insuficiencia renal grave. Se estudió la farmacocinética de ledipasvir, sofosbuvir y GS-331007 en pacientes infectados por el VHC con NT que requerían diálisis tratados con ledipasvir/sofosbuvir (n = 94) durante 8, 12 o 24 semanas, y se comparó con pacientes sin insuficiencia renal en los ensayos de fase 2/3 de ledipasvir/sofosbuvir. Insuficiencia hepática: La farmacocinética de ledipasvir se estudió con una dosis única de 90 mg de ledipasvir en pacientes negativos para el VHC con insuficiencia hepática grave (clase C de Child-Pugh-Turcotte [CPT]). La exposición plasmática (AUCinf) a ledipasvir fue similar en los pacientes con insuficiencia hepática grave y en los pacientes con función hepática normal que actuaron como controles. Los análisis farmacocinéticos poblacionales en pacientes infectados por el VHC indicaron que la cirrosis (incluida la cirrosis descompensada) no tenía un efecto clínicamente relevante sobre la exposición a ledipasvir. La farmacocinética de sofosbuvir se estudió tras la administración de 400 mg del medicamento durante 7 días en pacientes infectados por el VHC con insuficiencia hepática moderada y grave (clases B y C de CPT). En comparación con los pacientes con función hepática normal, el AUC0-24 de sofosbuvir fue un 126 % y un 143 % más alta en los pacientes con insuficiencia hepática moderada y grave, mientras que el AUC0-24 de GS-331007 fue un 18 % y un 9 % más alta, respectivamente. Los análisis farmacocinéticos poblacionales en pacientes infectados por el VHC indicaron que la cirrosis (incluida la cirrosis descompensada) no tenía un efecto clínicamente relevante sobre la exposición a sofosbuvir y a GS-331007. Peso corporal: El peso corporal no tuvo un efecto significativo sobre la exposición a sofosbuvir según un análisis farmacocinético poblacional. La exposición a ledipasvir disminuye con el aumento del peso corporal pero el efecto no se considera clínicamente relevante. Población pediátrica: Las exposiciones a ledipasvir, sofosbuvir y GS-331007 en adolescentes de 12 a < 18 años de edad fueron similares a las de los adultos participantes en los estudios de fases 2/3 tras la administración de ledipasvir/sofosbuvir (90 mg/400 mg). No se ha establecido la farmacocinética de ledipasvir, sofosbuvir y de GS-331007 en los pacientes pediátricos menores de 12 años (ver sección 4). Microbiología: Actividad antiviral: Los valores de CE50 de ledipasvir y sofosbuvir frente a replicones de longitud completa o quiméricos capaces de codificar secuencias de la NS5A y NS5B a partir de aislamientos clínicos se detallan en la Tabla 2. La presencia de suero humano al 40 % no tuvo efectos sobre la actividad de sofosbuvir contra el VHC pero redujo 12 veces la actividad de ledipasvir contra el VHC frente a los replicones del VHC de genotipo 1a. (Ver Tabla 2).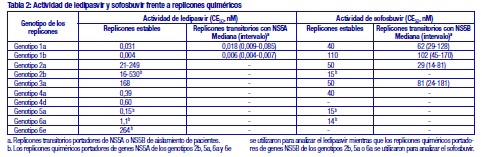
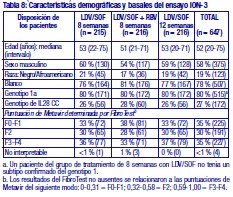
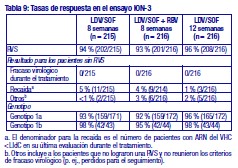
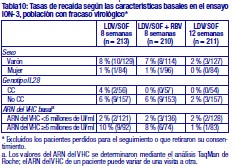
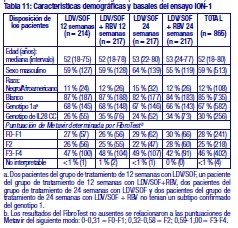
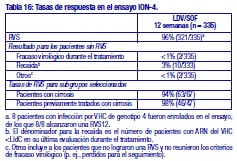
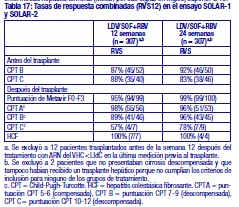
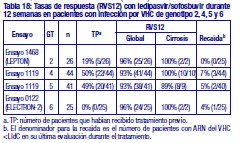
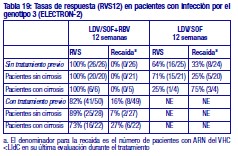
Resistencia: En cultivos celulares: Se han seleccionado replicones del VHC con sensibilidad reducida a ledipasvir en cultivos celulares para los genotipos 1a y 1b. La sensibilidad reducida a ledipasvir se asoció con la sustitución primaria de la NS5A Y93H en ambos genotipos, 1a y 1b. Además, apareció una sustitución Q30E en los replicones de genotipo 1a. La mutagénesis dirigida al sitio de las VAR de la NS5A mostró que las sustituciones que confieren una magnitud del cambio > 100 y ≤1.000 de la sensibilidad a ledipasvir son Q30H/R, L31I/M/V, P32L e Y93T en el genotipo 1a y P58D y Y93S en el genotipo 1b; y las sustituciones que confieren una magnitud del cambio > 1.000 son M28A/G, Q30E/G/K, H58D, Y93C/H/N/S en el genotipo 1a y A92K e Y93H en el genotipo 1b. Se han seleccionado replicones del VHC con sensibilidad reducida a sofosbuvir en cultivos celulares para múltiples genotipos, entre ellos 1b, 2a, 2b, 3a, 4a, 5a y 6a. La sensibilidad reducida a sofosbuvir se asoció con la sustitución primaria de la NS5B S282T en todos los genotipos de replicón estudiados. La mutagénesis dirigida al sitio de la sustitución S282T en los replicones de 8 genotipos redujo la sensibilidad a sofosbuvir de 2 a 18 veces y disminuyó la capacidad de replicación viral en un 89 % a 99 % en comparación con el tipo salvaje correspondiente. En ensayos clínicos - Adultos-genotipo 1: En un análisis combinado de pacientes que en los ensayos de fase 3 (ION-3, ION-1 e ION-2) recibieron ledipasvir/sofosbuvir, 37 (29 con genotipo 1a y 8 con genotipo 1b) reunían los requisitos necesarios para los análisis de resistencia debido a que presentaron fracaso virológico o interrupción precoz del fármaco del ensayo y un ARN del VHC > 1.000 UI/ml. Se disponía de datos de secuenciación profunda (punto de corte de la prueba: 1 %) de la NS5A y la NS5B posteriores a la situación basal para 37/37 y 36/37 pacientes, respectivamente. Se observaron variantes asociadas a resistencia (VAR) de la NS5A en aislamientos posteriores a la situación basal de 29/37 pacientes (22/29 de genotipo 1a y 7/8 de genotipo 1b) que no alcanzaron una respuesta virológica sostenida (RVS). De los 29 pacientes con genotipo 1a que reunían los requisitos para los análisis de resistencia, 22/29 (76 %) pacientes albergaban una o más VAR de la NS5A en las posiciones K24, M28, Q30, L31, S38 e Y93 en el momento del fracaso, mientras que en los 7/29 pacientes restantes no se detectaron VAR de la NS5A en el momento del fracaso. Las variantes más frecuentes fueron Q30R, Y93H y L31M. De los 8 pacientes con genotipo 1b que reunían los requisitos para los análisis de resistencia, 7/8 (88 %) albergaban una o más VAR de la NS5A en las posiciones L31 e Y93 en el momento del fracaso, mientras que 1/8 pacientes carecía de VAR de la NS5A en el momento del fracaso. La variante más frecuente fue Y93H. Entre los 8 pacientes que carecían de VAR de la NS5A en el momento del fracaso, 7 recibieron 8 semanas de tratamiento (n = 3 con ledipasvir/sofosbuvir; n = 4 con ledipasvir/sofosbuvir + ribavirina) y 1 paciente recibió ledipasvir/sofosbuvir durante 12 semanas. En los análisis fenotípicos, los aislamientos posteriores a la situación basal de los pacientes que albergaban VAR de la NS5A en el momento del fracaso mostraron una sensibilidad reducida de 20 al menos 243 veces (la dosis más alta analizada) a ledipasvir. La mutagénesis dirigida al sitio de la sustitución Y93H en ambos genotipos, 1a y 1b, y las sustituciones Q30R y L31M en el genotipo 1a redujeron en gran medida la sensibilidad a ledipasvir (magnitud del cambio en la CE50 que oscilaba entre 544 y 1.677 veces). Entre los pacientes con hepatopatía compensada después del trasplante o los pacientes con hepatopatía descompensada antes o después del trasplante (ensayos SOLAR-1 y SOLAR-2), la recaída se asoció con la detección de uno o más de los siguientes VAR de la NS5A: K24R, M28T, Q30R/H/K, L31V, H58D e Y93H/C en 12/14 pacientes de genotipo 1a, y L31M, Y93H/N en 6/6 pacientes de genotipo 1b. La sustitución E237G de la NS5B se detectó en 3 pacientes (1 de genotipo 1b y 2 de genotipo 1a) en los ensayos de fase 3 (ION-3, ION-1 e ION-2) y en 3 pacientes con infección por el genotipo 1a en los ensayos SOLAR-1 y SOLAR-2 en el momento de la recaída. La sustitución E237G mostró una reducción de 1,3 veces en la sensibilidad al sofosbuvir en el ensayo de replicones de genotipo 1a. La trascendencia clínica de esta sustitución se desconoce actualmente. La sustitución S282T de la NS5B asociada con resistencia a sofosbuvir no se detectó en ningún aislamiento de fracaso virológico de los ensayos de fase 3. Sin embargo, la sustitución S282T de la NS5B, en combinación con las sustituciones de la NS5A L31M, Y93H y Q30L, se detectó en un paciente en el momento del fracaso tras 8 semanas de tratamiento con ledipasvir/sofosbuvir en un ensayo de fase 2 (LONESTAR). Este paciente se volvió a tratar posteriormente con ledipasvir/sofosbuvir + ribavirina durante 24 semanas y alcanzó una RVS tras dicha repetición del tratamiento. En el ensayo SIRIUS (ver "Eficacia clínica y seguridad" más abajo), 5 pacientes con infección por el genotipo 1 recayeron después del tratamiento con ledipasvir/sofosbuvir con o sin ribavirina. Se observaron VAR de la NS5A en la recaída en 5/5 pacientes (para el genotipo 1a: Q30R/H + L31M/V [n = 1] y Q30R [n = 1]; para el genotipo 1b: Y93H [n = 3]). En ensayos clínicos - Adultos-genotipo 2, 3, 4, 5 y 6: VAR de la NS5A: Ninguno de los pacientes infectados por el genotipo 2 experimentaron recaída en el ensayo clínico, por lo que no se dispone de datos relativos a las VAR de la NS5A en el momento del fracaso. En los pacientes infectados por el genotipo 3 que experimentaron fracaso virológico, no se detectó de forma típica el desarrollo de VAR de la NS5A (incluido el incremento de las VAR presentes en la situación basal) en el momento del fracaso (n = 17). En cuanto a la infección por el genotipo 4, 5 y 6, solo se ha evaluado un número escaso de pacientes (un total de 5 pacientes con fracaso). La sustitución Y93C de la NS5A apareció en el VHC de 1 paciente (genotipo 4), mientras que las VAR de la NS5A presentes en la situación basal se observaron en el momento del fracaso en todos los pacientes. En el ensayo SOLAR-2, un paciente con genotipo 4d presentó sustitución E237G de la NS5B en el momento de la recaída. La trascendencia clínica de esta sustitución se desconoce actualmente. VAR de la NS5B: La sustitución S282T de la NS5B apareció en el VHC de 1/17 fracasos en el genotipo 3 y en el VHC de 1/3, 1/1 y 1/1 fracasos en el genotipo 4, 5 y 6, respectivamente. Efecto de las variantes basales del VHC asociadas con resistencia sobre el resultado del tratamiento: Adultos-Genotipo 1: Se realizaron análisis para investigar la asociación entre las VAR de la NS5A basales preexistentes y el resultado del tratamiento. En el análisis combinado de los ensayos de fase 3, un 16 % de los pacientes presentaba VAR de la NS5A basales identificadas mediante secuenciación poblacional o profunda, independientemente del subtipo. Las VAR de la NS5A basales estaban sobrerrepresentadas en pacientes que experimentaron recaída en los ensayos de fase 3 (ver "Eficacia clínica y seguridad"). Tras 12 semanas de tratamiento con ledipasvir/sofosbuvir (sin ribavirina) en pacientes con tratamiento previo (grupo 1 del estudio ION-2) 4/4 pacientes con una VAR de la NS5A basal que confiere una magnitud del cambio en la resistencia a ledipasvir de ≤100 alcanzaron una RVS. Para el mismo grupo de tratamiento, en los pacientes con VAR de la NS5A basal que confieren una magnitud del cambio de > 100 se produjo recaída en 4/13 (31 %), en comparación con 3/95 (3 %) en los que no tenían ninguna VAR basal o con VAR que confieren una magnitud del cambio de ≤100. Tras 12 semanas de tratamiento con ledipasvir/sofosbuvir con ribavirina en pacientes con tratamiento previo y cirrosis compensada (SIRIUS, n = 77), 8/8 pacientes con VAR de la NS5A basales que confieren una sensibilidad reducida > 100 veces al ledipasvir alcanzaron una RVS12. Entre los pacientes con hepatopatía compensada después del trasplante (ensayos SOLAR-1 y SOLAR-2) no se produjo recaída en los pacientes con VAR de la NS5A basal (n = 23) tras 12 semanas de tratamiento con ledipasvir/sofosbuvir con ribavirina. Entre los pacientes con hepatopatía descompensada (antes y después del trasplante), en 4/16 pacientes (el 25 %) con VAR de la NS5A que confieren una resistencia > 100 veces se produjo recaída tras 12 semanas de tratamiento con ledipasvir/sofosbuvir con ribavirina comparado con 7/120 (el 6 %) en los que no tenían ninguna VAR de la NS5A basal o con VAR que confieren una magnitud del cambio de ≤100. El grupo de VAR de la NS5A que confirió un cambio > 100 veces y se observó en los pacientes fueron las siguientes sustituciones en el genotipo 1a (M28A, Q30H/R/E, L31M/V/I, H58D, Y93H/N/C) o en el genotipo 1b (Y93H). La proporción de dichas VAR de la NS5A basales observadas con secuenciación profunda osciló entre muy baja (punto de corte de la prueba = 1 %) y alta (parte principal de la población del plasma). La sustitución asociada con resistencia a sofosbuvir S282T no se detectó en la secuencia basal de la NS5B de ningún paciente de los ensayos de fase 3 mediante secuenciación poblacional o profunda. Se alcanzó una RVS en los 24 pacientes (n = 20 con L159F + C316N; n = 1 con L159F; y n = 3 con N142T) que presentaban variantes basales asociadas con resistencia a los inhibidores nucleosídicos de NS5B. Adultos-Genotipo 2, 3, 4, 5 y 6: Debido al limitado tamaño de los ensayos, no se ha evaluado completamente la repercusión de las VAR de la NS5A basales sobre el resultado del tratamiento en los pacientes con HCC de genotipo 2, 3, 4, 5 o 6. No se observaron diferencias importantes en los resultados en función de la presencia o ausencia de VAR de la NS5A basales. Resistencia cruzada: Ledipasvir fue totalmente activo frente a la sustitución asociada con resistencia a sofosbuvir S282T de la NS5B, mientras que todas las sustituciones asociadas con resistencia a ledipasvir de la NS5A fueron totalmente sensibles a sofosbuvir. Tanto sofosbuvir como ledipasvir fueron totalmente activos frente a las sustituciones asociadas con resistencia a otras clases de antivirales de acción directa con diferentes mecanismos de acción, como los inhibidores no nucleósidos de la NS5B y los inhibidores de la proteasa NS3. Las sustituciones de la NS5A que confieren resistencia a ledipasvir pueden reducir la actividad antiviral de otros inhibidores de NS5A.
Indicaciones.
HARVONI® está indicado para el tratamiento de la hepatitis C crónica (HCC) en adultos y en adolescentes entre 12 y 18 años de edad. Para consultar la actividad específica frente al genotipo del virus de la hepatitis C (VHC), ver secciones Advertencias - Generales, Mecanismo de acción, Microbiología. y Estudios clínicos.
Dosificación.
El tratamiento con HARVONI® debe ser iniciado y controlado por un médico con experiencia en el tratamiento de los pacientes con HCC. Posología: Adultos y adolescentes entre 12 y 18 años de edad: La dosis recomendada de HARVONI® es de un comprimido una vez al día, acompañado o no de alimentos (ver sección 3.2.2).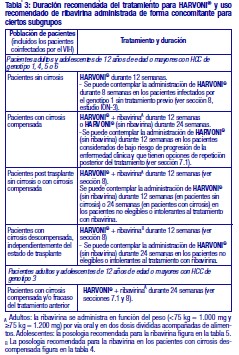

Cuando HARVONI® se utilice en combinación con ribavirina, consulte también la Información de Prescripción de la ribavirina. En pacientes adolescentes de 12 a 18 años de edad se recomienda la siguiente posología para la ribavirina cuando se divida en dos dosis diarias y se administre con alimentos:
Modificación de la dosis de ribavirina en adultos que toman 1.000-1.200 mg al día: Si se utiliza HARVONI® en combinación con ribavirina y un paciente presenta una reacción adversa grave potencialmente relacionada con ribavirina, la dosis de ribavirina se debe modificar o interrumpir, si es pertinente, hasta que la reacción adversa remita o disminuya su gravedad. En la Tabla 6 se facilitan las directrices de modificación e interrupción de la dosis en función de la concentración de hemoglobina y el estado cardiaco del paciente.
Una vez suspendida ribavirina a causa de una anomalía analítica o una manifestación clínica, se puede intentar reanudarla a una dosis de 600 mg al día y posteriormente incrementarla a 800 mg al día. Sin embargo, no se recomienda aumentarla a la dosis originalmente asignada (1.000 mg a 1.200 mg al día). Población pediátrica menores de 12 años de edad: No se ha establecido la seguridad y la eficacia de HARVONI® en niños menores de 12 años. No se dispone de datos en pacientes pediátricos menores de 12 años de edad. Dosis omitida: Se debe indicar a los pacientes que si vomitan en un plazo de 5 horas desde la administración, deben tomar un comprimido adicional. Si vomitan más de 5 horas después de la administración, no hace falta ninguna dosis adicional (ver sección 8). Si se omite una dosis y no han transcurrido 18 horas desde la hora normal, se debe indicar a los pacientes que tomen el comprimido lo antes posible y después los pacientes deben tomar la siguiente dosis a la hora habitual. Si han transcurrido más de 18 horas, se debe indicar entonces a los pacientes que esperen y tomen la siguiente dosis a la hora habitual. Se debe indicar a los pacientes que no tomen una dosis doble. Pacientes de edad avanzada: No es preciso ajustar la dosis en los pacientes de edad avanzada (ver sección Farmacocinética). Insuficiencia renal: No es necesario ajustar la dosis de HARVONI® en los pacientes con insuficiencia renal leve o moderada. Se dispone de datos muy limitados sobre la seguridad de ledipasvir/sofosbuvir en los pacientes con insuficiencia renal grave (tasa de filtración glomerular estimada [TFGe] < 30 ml/min/1,73 m²) (ver las secciones Farmacocinética y Estudios clínicos). No se ha evaluado la seguridad en pacientes con nefropatía terminal (NT) que precisa diálisis (ver la sección Estudios clínicos). El tratamiento con ledipasvir/sofosbuvir se debe considerar en pacientes con insuficiencia renal grave o NT solo cuando no se puedan utilizar tratamientos alternativos, recomendados para estos pacientes (ver sección Advertencias). Insuficiencia hepática: No es necesario ajustar la dosis de HARVONI® en los pacientes con insuficiencia hepática leve, moderada o grave (clases A, B o C de CPT, ver sección Farmacocinética). Se ha establecido la seguridad y eficacia de ledipasvir/sofosbuvir en los pacientes con cirrosis descompensada (ver sección Estudios clínicos). Población pediátrica: No se ha establecido la seguridad y la eficacia de HARVONI® en niños menores de 12 años. No se dispone de datos en pacientes pediátricos menores de 12 años de edad. Forma de administración: Por vía oral. Se debe indicar a los pacientes que traguen el comprimido entero, acompañado o no de alimentos. Debido a su sabor amargo, se recomienda no masticar ni machacar el comprimido recubierto (ver sección Farmacocinética). Formas farmacéuticas y concentraciones: Comprimido recubierto con película de color anaranjado y forma de rombo, con "GSI" grabado por un lado y "7985" por el otro. Cada comprimido recubierto contiene 90 mg de ledipasvir y 400 mg de sofosbuvir.
Contraindicaciones.
Hipersensibilidad a los principios activos o a alguno de los excipientes. La administración concomitante con rosuvastatina (ver sección Interacciones). Uso con inductores potentes de la glucoproteína P: Medicamentos que son inductores potentes de la glucoproteína P en el intestino (carbamacepina, fenofarbital, fenitoína, rifampicina, rifabutina y hierba de San Juan [Hypericum perforatum]). La administración concomitante reducirá significativamente las concentraciones plasmáticas de ledipasvir y sofosbuvir y puede provocar la disminución de la eficacia de HARVONI® (ver sección Interacciones).
Reacciones adversas.
Resumen del perfil de seguridad en adultos: La evaluación de la seguridad de ledipasvir/sofosbuvir se basó principalmente en los datos agrupados de tres ensayos clínicos de fase 3 sin control, en 1952 pacientes que recibieron ledipasvir/sofosbuvir durante 8, 12 o 24 semanas, incluyendo 872 pacientes que recibieron un tratamiento combinado de ledipasvir/sofosbuvir + ribavirina El porcentaje de pacientes que suspendieron permanentemente el tratamiento debido a acontecimientos adversos fue del 0 %, < 1 % y 1 % para los pacientes que recibieron ledipasvir/sofosbuvir durante 8, 12 y 24 semanas, respectivamente, y de < 1 %, 0 %, y 2 % para los pacientes que recibieron el tratamiento combinado de ledipasvir/sofosbuvir + ribavirina durante 8, 12 y 24 semanas, respectivamente. En los ensayos clínicos, la fatiga y la cefalea fueron más frecuentes en los pacientes tratados con ledipasvir/sofosbuvir en comparación con placebo. Cuando ledipasvir/sofosbuvir se estudió junto con ribavirina, las reacciones adversas más frecuentes del tratamiento combinado de ledipasvir/sofosbuvir + ribavirina concordaron con el perfil de seguridad conocido de la ribavirina, sin un aumento de la frecuencia ni de la gravedad de las reacciones adversas previstas. Se han identificado las siguientes reacciones adversas con HARVONI® (Tabla 20). Las reacciones adversas se incluyen a continuación según el sistema de clasificación de órganos y frecuencia. Las frecuencias se definen del siguiente modo: muy frecuentes (≥1/10), frecuentes (≤1/100 a < 1/10), poco frecuentes (≥1/1.000 a < 1/100), raras (≥1/10.000 a < 1/1.000) o muy raras ( < 1/10.000).
Adultos con cirrosis descompensada y/o que están a la espera de un trasplante hepático o después de un trasplante hepático: Se evaluó el perfil de seguridad de ledipasvir/sofosbuvir con ribavirina durante 12 o 24 semanas en adultos con hepatopatía descompensada y/o después de un trasplante hepático en dos ensayos abiertos (SOLAR-1 y SOLAR-2). No se detectaron reacciones adversas medicamentosas nuevas en los pacientes con cirrosis descompensada y/o después de un trasplante hepático que recibieron ledipasvir/sofosbuvir con ribavirina. Aunque se produjeron acontecimientos adversos, incluidos acontecimientos adversos graves, con mayor frecuencia en este ensayo que en los ensayos en los que se excluyó a los pacientes descompensados y/o después de un trasplante hepático, los acontecimientos adversos observados fueron los esperados como secuelas clínicas de una hepatopatía avanzada y/o un trasplante hepático o fueron consistentes con el perfil de seguridad conocido de la ribavirina (ver sección 8 para información detallada sobre este estudio). El 39% y el 13% de los pacientes tratados con ledipasvir/sofosbuvir con ribavirina presentaron disminuciones de la hemoglobina a < 10 g/dl y < 8,5 g/dl, respectivamente, durante el tratamiento. La ribavirina se interrumpió en el 15% de los pacientes. En el 7% de los receptores de trasplante hepático se realizó una modificación de los medicamentos inmunosupresores. Pacientes con insuficiencia renal: En un estudio clínico abierto (Estudio 0154) se administró ledipasvir/sofosbuvir durante 12 semanas a 18 pacientes con HCC de genotipo 1 e insuficiencia renal grave. En este conjunto limitado de datos sobre la seguridad clínica, la tasa de acontecimientos adversos no aumentó claramente con respecto a lo que se espera en pacientes con insuficiencia renal grave. La seguridad de HARVONI® se evaluó en un estudio no controlado de 12 semanas que incluyó a 95 pacientes con NT que requerían diálisis (Estudio 4063). En este contexto, la exposición al metabolito de sofosbuvir GS-331007 aumentó 20 veces, superando los niveles para los cuales se han observado reacciones adversas en ensayos preclínicos. En este conjunto limitado de datos sobre la seguridad clínica, la tasa de acontecimientos adversos y muertes no aumentó claramente con respecto a lo que se espera en pacientes con NT. Población pediátrica La seguridad y la eficacia de HARVONI® en niños y adolescentes de 12 a < de 18 años se basa en los datos obtenidos en un estudio clínico abierto de fase 2 (Estudio 1116) en el que participaron 100 pacientes con infección por el VHC de genotipo 1 que fueron tratados con ledipasvir/sofosbuvir durante 12 semanas. Las reacciones adversas observadas fueron coherentes con las constatadas en los estudios clínicos de ledipasvir/sofosbuvir en adultos (ver la tabla 1). Descripción de las reacciones adversas seleccionadas; Arritmias cardíacas: Se han observado casos de bradicardia severa y bloqueo cardiaco cuando HARVONI® se utiliza con amiodarona, con o sin otros fármacos antiarrítmicos (ver secciones Advertencias - Generales e Interacciones). Trastornos de la piel: Frecuencia no conocida: Síndrome de Stevens-Johnson Notificación de Sospecha de Reacciones Adversas: Es importante notificar la sospecha de reacciones adversas al medicamento tras su autorización. Ello permite una supervisión continuada de la relación beneficio/riesgo del medicamento. Se invita a los profesionales sanitarios a notificar las sospechas de reacciones adversas a través del Sistema Nacional de Farmacovigilancia al siguiente link: http://sistemas.anmat.gov.ar/aplicaciones_net/applications/fvg_eventos_adversos_nuevo/index.html y/o al Departamento de Farmacovigilancia de GADOR S.A. vía email a farmacovigilancia@gador.com.ar o telefónicamente al 0800-220-2273.
Advertencias.
Generales: HARVONI® no se debe administrar de forma concomitante con otros medicamentos que contengan sofosbuvir. Actividad específica según genotipo: En cuanto a las pautas recomendadas con diferentes genotipos del VHC, ver sección Dosificación. En cuanto a la actividad virológica y clínica específica según genotipo, ver sección Estudios clínicos. Los datos clínicos para respaldar el uso de HARVONI® en adultos infectados por el VHC de genotipo 3 son limitados (ver sección Estudios clínicos). No se ha investigado la eficacia relativa de una pauta de 12 semanas consistente en ledipasvir/sofosbuvir + ribavirina, comparada con una pauta de 24 semanas de sofosbuvir + ribavirina. Se recomienda un tratamiento conservador de 24 semanas en todos los pacientes de genotipo 3 con tratamiento previo y en los pacientes de genotipo 3 sin ningún tratamiento previo y con cirrosis (ver sección Dosificación). En la infección por el genotipo 3, solo se debe contemplar el uso de HARVONI® (siempre en combinación con ribavirina) en los pacientes considerados de alto riesgo de progresión clínica de la enfermedad y cuando no existan opciones terapéuticas alternativas. Los datos clínicos para respaldar el uso de HARVONI® en adultos infectados por el VHC de genotipo 2 y 6 son limitados (ver sección Estudios clínicos). Bradicardia severa y bloqueo cardiaco: Se han observado casos de bradicardia severa y bloqueo cardiaco cuando HARVONI® se utiliza con amiodarona, con o sin otros fármacos para disminuir la frecuencia cardiaca. El mecanismo no está establecido. El uso concomitante de amiodarona fue limitado durante el desarrollo clínico de sofosbuvir. Los casos son potencialmente mortales, por lo que la amiodarona solo se debe administrar a pacientes que toman HARVONI® cuando no se toleren o estén contraindicados otros tratamientos antiarrítmicos. Si el uso concomitante de amiodarona se considera necesario, se recomienda una estrecha vigilancia de los pacientes cuando se inicie la administración de HARVONI®. Los pacientes de alto riesgo de bradiarritmia se deben monitorizar de forma continua durante 48 horas en un entorno clínico adecuado. Debido a la prolongada semivida de la amiodarona, también se deben monitorizar adecuadamente aquellos pacientes que hayan dejado de tomar amiodarona pocos meses antes y vayan a comenzar el tratamiento con HARVONI®. A todos los pacientes que reciben HARVONI® en combinación con amiodarona, con o sin otros fármacos antiarrítmicos, se les debe indicar cuáles son los síntomas de bradicardia y bloqueo cardiaco, e indicarles que acudan urgentemente al médico si experimentan dichos síntomas. Uso en pacientes diabéticos: Los diabéticos pueden experimentar un mejor control de la glucosa, lo que puede resultar en hipoglucemia sintomática, después de inicia