NOVOEIGHT®
NOVO NORDISK
Grupo farmacoterapéutico: Antihemorrágico, factor VIII de coagulación sanguínea. Código ATC: B02BD02.
Composición.
Cada vial de polvo contiene nominalmente 250, 500, 1000, 1500, 2000 o 3000 UI de factor VIII recombinante humano (ADNr), turoctocog alfa. Luego de la reconstitución, NovoEight® contiene aproximadamente 62,5, 125, 250, 375, 500 o 750 UI/ml de factor VIII recombinante humano (ADNr), turoctocog alfa. La potencia (UI) se determina utilizando el ensayo cromogénico de Farmacopea Europea (Ph. Eur.). La actividad específica de NovoEight® es de aproximadamente 8,300 UI/mg de proteína. Turoctocog alfa (factor VIII recombinante humano (ADNr)) es una proteína purificada que contiene 1445 aminoácidos con una masa molecular de aproximadamente 166 kDA. Se produce utilizando tecnología de ADN recombinante en células de Ovario de Hámster Chino (CHO) y preparado sin la adición de derivados proteicos humanos o animales en el proceso de cultivo, purificación o formulación final. Turoctocog alfa es un factor VIII de coagulación humano recombinante truncado en el dominio B (el dominio B consiste en 21 aminoácidos del dominio B natural) sin ninguna otra modificación de la secuencia de aminoácidos. Excipientes: Polvo Liofilizado: Cloruro de sodio, L-histidina, Sacarosa, Polisorbato 80, L-metionina, Cloruro de calcio dihidratado, Hidróxido de sodio, Ácido clorhídrico; Solvente: Cloruro de sodio, Agua para inyectables. Excipiente con efecto conocido: Este medicamento contiene 30,5 mg de sodio por vial reconstituido Forma farmacéutica: Polvo liofilizado y solvente para solución inyectable. Polvo o masa friable de color blanco o ligeramente amarillo. Solución inyectable transparente e incolora.
Farmacología.
Propiedades farmacodinámicas: Mecanismo de acción: NovoEight® contiene turoctocog alfa, un factor VIII recombinante humano (ADNr) con un dominio B truncado. Esta glicoproteína tiene la misma estructura que el factor VIII humano cuando se activa, y las modificaciones post-traduccionales son similares a aquellas en la molécula derivada del plasma. Se ha observado que el punto de sulfatación de tirosina presente en Tyr1680 (longitud completa nativa), que es importante para el enlace con el factor de von Willebrand, está totalmente sulfatado en la molécula de turoctocog alfa. Cuando se administra en infusión en pacientes hemofílicos, el factor VIII se une al Factor de von Willebrand endógeno en la circulación del paciente. El complejo factor VIII/factor de von Willebrand consiste en dos moléculas (factor VIII y Factor de von Willebrand) con diferentes funciones fisiológicas. El factor VIII activado actúa como un cofactor para el factor IX activado, acelerando la conversión del factor X a factor X activado. El factor X activado transforma la protrombina en trombina. La trombina, luego, convierte el fibrinógeno en fibrina y se puede formar el coágulo. La hemofilia A es un desorden hereditario de la coagulación sanguínea vinculado al sexo provocado por los niveles disminuidos del factor VIII:C lo que resulta en sangrados profusos en las articulaciones, músculos u órganos internos, ya sea espontáneamente o como resultado de un trauma accidental o quirúrgico. Mediante una terapia de sustitución los niveles plasmáticos del factor VIII se incrementan, logrando una corrección temporal de la deficiencia del factor y, por consiguiente, la corrección de la tendencia al sangrado. A destacar, no se puede comparar la tasa anual de hemorragia (ABR, por sus siglas en inglés) entre los diferentes concentrados de factores y entre los diferentes estudios clínicos. Datos de eficacia clínica y seguridad: Se llevaron a cabo cuatro estudios multicéntricos, abiertos, no controlados para evaluar la seguridad y eficacia de NovoEight® en la prevención y tratamiento de sangrados, y durante cirugía en pacientes con hemofilia A severa (Actividad Factor VIII ≤1 %). Tres de estos estudios se realizaron en pacientes tratados previamente y el cuarto en pacientes no tratados previamente. Los estudios incluyeron 298 pacientes expuestos; 175 adolescentes o adultos sin inhibidores con edades desde los 12 años (150 días de exposición), 63 pacientes pediátricos tratados previamente sin inhibidores menores de 12 años (50 días de exposición) y 60 pacientes no tratados previamente menores de 6 años de edad. Continuaron en el estudio de extensión de seguridad 188 de los 238 pacientes tratados previamente. El tratamiento con NovoEight® mostró ser seguro y tener el efecto hemostático y preventivo deseado. De los 3293 sangrados reportados en 298 pacientes, 2902 de ellos (88,1%) se resolvieron con 1-2 infusiones de NovoEight®.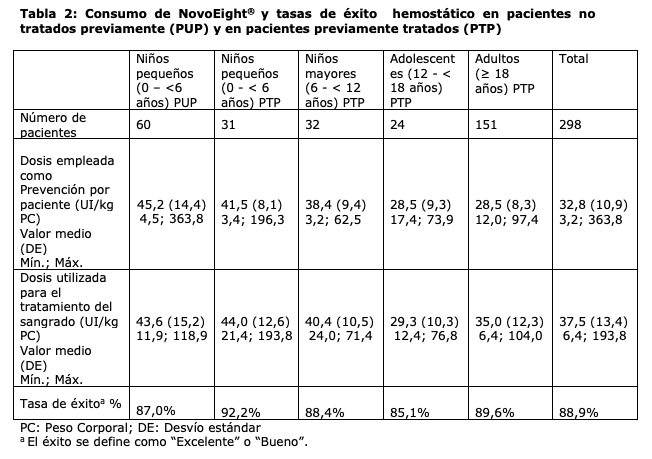
Se llevaron a cabo un total de 30 cirugías en 25 pacientes de las cuales 26 fueron cirugías mayores y 4 cirugías menores. La hemostasia fue exitosa en todas las cirugías y no se reportaron fallas en el tratamiento. Se han recopilado datos de la Inducción de la Tolerancia Inmune (ITI) en pacientes con hemofilia A que han desarrollado inhibidores del factor VIII. Durante los estudios clínicos en PUP, se trataron a 21 pacientes con ITI y 18 (86%) pacientes completaron la ITI con un resultado negativo para la prueba de detección de inhibidores. Propiedades farmacocinéticas: Todos los estudios farmacocinéticos (PK) con NovoEight® se llevaron a cabo tras la administración i.v de 50 IU/kg de NovoEight® en pacientes con hemofilia A severa tratados previamente (FVIII ≤ 1%). El análisis de las muestras plasmáticas se llevó a cabo usando tanto el ensayo de coagulación en una etapa, como el ensayo cromogénico. Se evaluó el rendimiento de NovoEight® en ensayos de FVIII:C y se comparó con un medicamento comercializado de FVIII recombinante de longitud completa. El estudio reveló que se obtienen resultados comparables y coherentes con ambos productos y que NovoEight® se puede medir en plasma de forma fiable sin necesidad de usar un estándar aparte para NovoEight®. Los parámetros farmacocinéticos de las administraciones de dosis únicas de NovoEight® se listan en la Tabla 3 para el Ensayo de coagulación de una sola fase y en la Tabla 4 para el Ensayo cromogénico.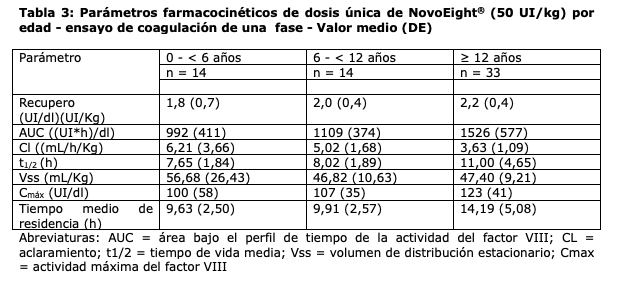
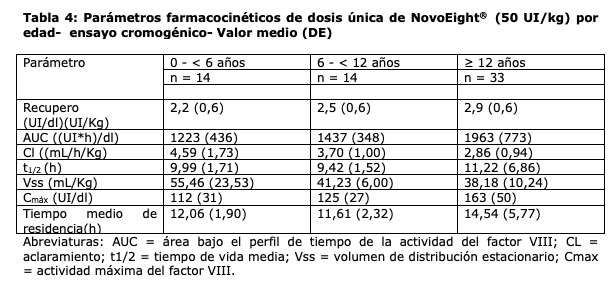
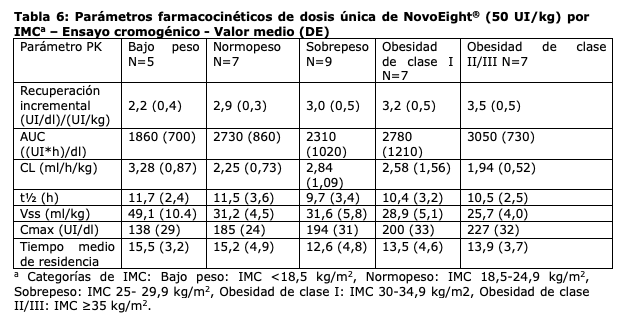
d máxima del factor VIII. Los parámetros farmacocinéticos fueron comparables entre los pacientes pediátricos por debajo de los 6 años y los pacientes entre 6 años y menos de 12 años de edad. Se observaron algunas variaciones en los parámetros farmacocinéticos de NovoEight® entre los pacientes adultos y los pediátricos. El mayor aclaramiento y la menor t1/2 vistos en pacientes pediátricos en comparación con los pacientes adultos con hemofilia A pueden deberse en parte al mayor volumen de plasma por kilogramo de peso corporal en los pacientes más jóvenes. Se realizó un estudio de parámetros farmacocinéticos de dosis única (50 UI/kg) en 35 pacientes con hemofilia (≥18 años) en diferentes categorías de Índice de Masa Corporal (IMC). La exposición máxima (Cmax) y la exposición total (AUC) se incrementaron al aumentarse el IMC lo que indica que puede ser necesario un ajuste de dosis en pacientes con bajo peso (IMC < 18,5 kg/m2) y en pacientes con obesidad (IMC ≥30 kg/m2), ver sección "Posología y modo de administración". 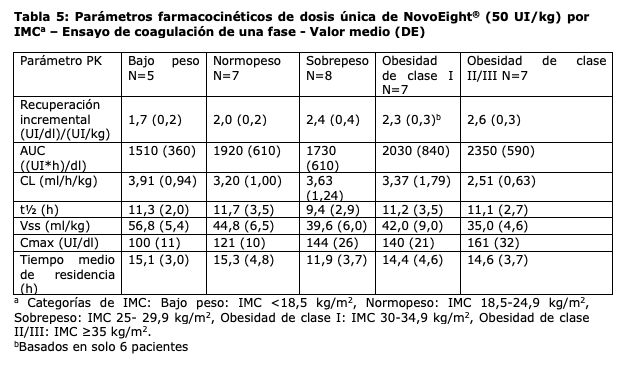
Datos preclínicos de seguridad: Los datos de los estudios no clínicos no muestran riesgos especiales en humanos basados en estudios convencionales de seguridad farmacológica y de toxicidad a dosis repetidas.
Indicaciones.
Tratamiento y profilaxis del sangrado para pacientes con hemofilia A (deficiencia congénita del factor VIII). NovoEight® puede ser utilizado en todos los grupos de edad.
Dosificación.
El tratamiento debe realizarse bajo la supervisión de un médico experto en el tratamiento de hemofilia. Supervisión del tratamiento: Durante el tratamiento, se recomienda una determinación apropiada de los niveles de factor VIII para orientar la dosis a administrar y la frecuencia de las inyecciones repetidas. La respuesta individual de cada paciente frente al factor VIII puede variar, demostrando diferentes vidas medias y recuperaciones. La dosis en función del peso corporal puede requerir un ajuste en pacientes con un peso por debajo del normal o con sobrepeso. En un estudio de parámetros farmacocinéticos de una sola dosis en adultos se incrementó la exposición máxima (Cmax) y la exposición total (AUC) al incrementarse el índice de masa corporal (IMC) lo que indica que puede ser necesario un ajuste de dosis. Se puede requerir un aumento de dosis en pacientes con bajo peso (IMC < 18,5 kg/m2) y se puede requerir una disminución de la dosis en pacientes con obesidad (IMC ≥30 kg/m2), no hay datos suficientes para recomendar ajustes de dosis específicos, ver sección "Propiedades farmacocinéticas". Especialmente en el caso de las intervenciones de cirugía mayor, es indispensable controlar con precisión la terapia de sustitución mediante pruebas de coagulación (actividad plasmática del factor VIII). Cuando se utiliza un ensayo in vitro de coagulación de tiempo de tromboplastina parcial activada (TTPa) para determinar la actividad del factor VIII en muestras de sangre de pacientes, los resultados de actividad del factor VIII del plasma pueden estar afectados significativamente tanto por el tipo de reactivo del TTPa como por el estándar de referencia usado en el ensayo. También, pueden ser significativas las discrepancias entre los resultados del ensayo obtenidos por el ensayo de coagulación de TTPa y el ensayo cromogénico según la Ph. Eur. Esto es de particular importancia cuando se cambia de laboratorio y/o los reactivos utilizados en el ensayo. Posología: La dosis y duración de la terapia de sustitución dependerá de la severidad de la deficiencia del factor VIII, de la localización y la extensión del sangrado y de la condición clínica del paciente. El número de unidades del factor VIII administrado se expresa como Unidades Internacionales (UI), que se relacionan con el estándar de la OMS vigente para productos de factor VIII. La actividad plasmática de factor VIII es expresada ya sea como porcentaje (con relación al nivel plasmático normal en humanos) o en Unidades Internacionales (con relación al Estándar Internacional de factor VIII en plasma). Una UI de actividad de Factor VIII es equivalente a la cantidad de Factor VIII en un mL de plasma humano normal. Tratamiento a demanda: El cálculo de la dosis requerida de Factor VIII se basa en los hallazgos empíricos de que 1 UI de factor VIII por Kg de peso corporal genera un aumento en la actividad plasmática del factor VIII de 2 UI/dL. La dosis requerida se determina usando la siguiente fórmula: Unidades requeridas (UI) = peso corporal (Kg) x aumento deseado de factor VIII (%) (UI/dL) x 0,5 (UI/Kg por UI/dL) La cantidad a administrarse y la frecuencia de la administración siempre deben orientarse a alcanzar la efectividad clínica en cada caso individual. En caso de los eventos hemorrágicos siguientes, la actividad del factor VIII no debe caer por debajo del nivel de actividad plasmática dada (en % de lo normal o en UI/dL) en el periodo correspondiente. Se puede usar la siguiente tabla para guiar la dosificación en los episodios de sangrado y en cirugía: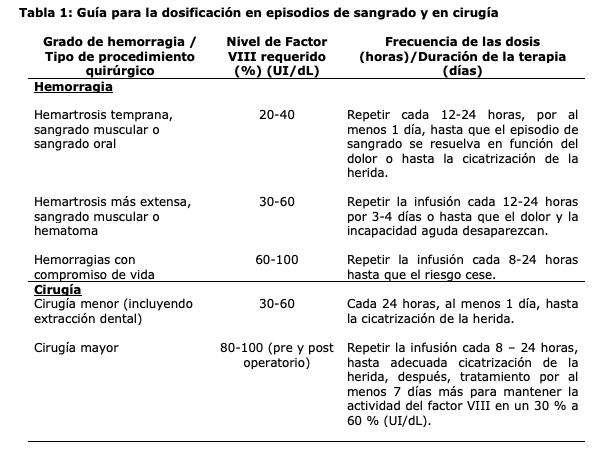
Profilaxis: En profilaxis a largo plazo para prevenir episodios de sangrado en pacientes con hemofilia A severa, las dosis recomendadas usualmente son de 20-40 UI de factor VIII por Kg de peso corporal cada dos días o 20-50 UI de factor VIII por Kg de peso corporal 3 veces a la semana. En adultos y adolescentes (mayores de 12 años) se puede aplicar un régimen menos frecuente (40-60 UI/kg cada 3 días o dos veces por semana). En algunos casos, especialmente en pacientes más jóvenes, pueden ser necesarios intervalos de dosis más cortos o dosis más elevadas. Cirugía: Existe experiencia limitada en cirugía en pacientes pediátricos. Personas de edad avanzada: No se tiene experiencia en pacientes > 65 años. Población pediátrica: En profilaxis a largo plazo para prevenir episodios de sangrado en pacientes menores de 12 años, se recomiendan dosis de 25-50 UI de Factor VIII por Kg de peso corporal cada dos días o de 25-60 UI de Factor VIII por Kg de peso corporal 3 veces a la semana. Para pacientes pediátricos mayores de 12 años la dosis recomendada es la misma que para adultos. Modo de administración: Vía intravenosa. La velocidad de infusión recomendada para NovoEight® es 1-2 ml/min. La velocidad debe determinarse según el nivel de comodidad del paciente. Para instrucciones sobre la reconstitución del producto antes de la administración, ver la sección Instrucciones sobre cómo usar NovoEight®.
Contraindicaciones.
Hipersensibilidad al principio activo o a cualquiera de los excipientes, listados en "Composición". Reacción alérgica conocida a proteínas de hámster.
Reacciones adversas.
Resumen del perfil de seguridad: Hipersensibilidad o reacciones alérgicas (que pueden incluir angioedema, ardor y punzadas en el punto de infusión, escalofríos, rubefacción, urticaria localizada o generalizada, cefalea,, hipotensión, letargia, náuseas, inquietud, taquicardia, opresión en el pecho, hormigueo, vómitos, sibilancias) se han observado raramente y, en algunos casos, puede progresar hasta una anafilaxia grave (incluido shock). En muy raras ocasiones se ha observado el desarrollo de anticuerpos frente a la proteína de hámster con reacciones de hipersensibilidad asociadas. Los pacientes con hemofilia A, tratados con factor VIII, incluido NovoEight® pueden desarrollar anticuerpos neutralizantes (inhibidores). Si se generan inhibidores de este tipo, la situación se pondrá de manifiesto como una respuesta clínica insuficiente. En tales casos, se recomienda ponerse en contacto con un centro especializado en hemofilia. Tabla de reacciones adversas: La tabla que se muestra a continuación sigue la clasificación de sistemas de órganos de MedDRA (clasificación por órganos y sistemas (SOC) y nivel de término preferente). Las categorías de frecuencia se definieron de acuerdo con la siguiente convención: muy frecuente (≥1/10), frecuente (≥1/100 a < 1/10), poco frecuente (≥1/1.000 a < 1/100), rara (≥1/10.000 a < 1/1.000), muy rara ( < 1/10.000), no conocida (no pueden ser estimadas con la información disponible). Dentro de cada grupo de frecuencia, las reacciones adversas se presentaron en orden decreciente de seriedad.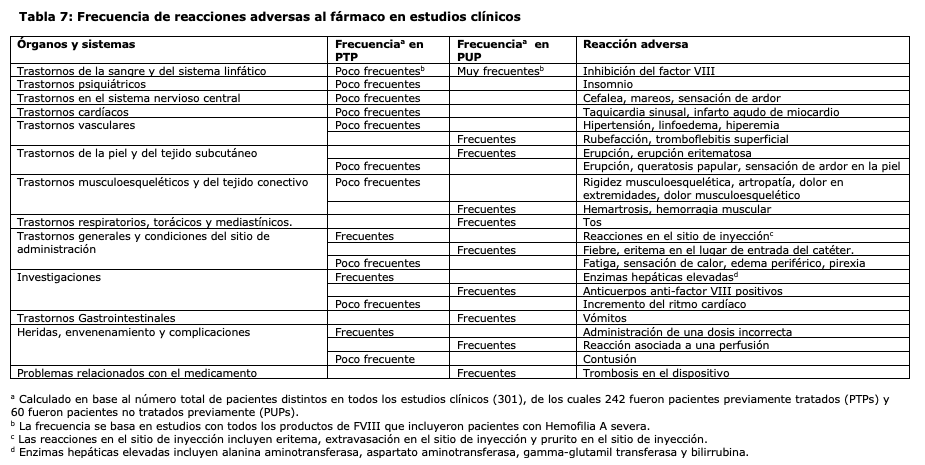
Descripción de las reacciones adversas seleccionadas: Durante todos los estudios clínicos con NovoEight® en pacientes tratados previamente, se han notificado un total de 35 reacciones adversas en 23 de 242 pacientes expuestos a NovoEight®. Las reacciones adversas que se informaron con mayor frecuencia fueron reacciones en el sitio de inyección, administración de una dosis incorrecta y enzimas hepáticas elevadas. De las 35 reacciones adversas, 2 de ellas se notificaron en 1 de 31 pacientes de menos de 6 años de edad, ninguna entre los pacientes de 6 a ≤12 años de edad, 1 reacción en 1 de 24 pacientes (de 12 a < 18 años de edad) y 32 en 21 de 155 adultos (≥18 años). Población pediátrica: En estudios clínicos que incluyeron 63 pacientes pediátricos tratados previamente entre 0 y 12 años de edad y 24 adolescentes entre 12 y 18 años, con hemofilia A severa, no se observó diferencia en el perfil de seguridad de NovoEight® entre los pacientes pediátricos y adultos. En el estudio con pacientes no tratados previamente, de entre 0 y 6 años de edad, se notificaron un total de 46 reacciones adversas en 33 de 60 pacientes expuestos a NovoEight®. La reacción adversa que se informó con mayor frecuencia fue la inhibición del Factor VIII, ver sección Advertencias y Precauciones Especiales de Uso. Se identificaron mutaciones genéticas de alto riesgo en el 92,3% del total y en el 93,8% de los que desarrollaron inhibidores confirmados con título elevado. Ningún otro factor se asoció de manera significativa con el desarrollo de inhibidores.
Advertencias.
Hipersensibilidad: Las reacciones de hipersensibilidad de tipo alérgico son posibles con NovoEight®. El producto contiene trazas de proteínas de hámster, lo cual puede generar reacciones alérgicas en algunos pacientes. Si se presentan síntomas de hipersensibilidad, se debe advertir a los pacientes de discontinuar el uso del medicamento inmediatamente y contactar a su médico. Los pacientes deben ser informados sobre los primeros signos de las reacciones de hipersensibilidad incluyendo urticaria localizada o generalizada, opresión en el pecho, sibilancias, hipotensión y anafilaxia. En caso de shock, se debe implementar el tratamiento médico estándar para shock. Inhibidores: La formación de anticuerpos neutralizantes (inhibidores) del factor VIII es una complicación conocida en el manejo de individuos con hemofilia A. Estos inhibidores son usualmente inmunoglobulinas IgG dirigidas contra la actividad procoagulante del factor VIII, y se cuantifican en Unidades Bethesda (BU) por mL de plasma utilizando la prueba modificada. El riesgo de desarrollar inhibidores se correlaciona con la severidad de la enfermedad, así como con la exposición al factor VIII; este riesgo es máximo dentro de los primeros 50 días de exposición, pero continúa durante toda la vida, aunque el riesgo es poco frecuente. La relevancia clínica del desarrollo de inhibidores dependerá del título del inhibidor, a saber: un título bajo presenta un menor riesgo de obtener una respuesta clínica insuficiente que un título alto de inhibidores. En general, todos los pacientes tratados con medicamentos de factor VIII de coagulación deben ser monitoreados cuidadosamente respecto al desarrollo de inhibidores por medio de una observación clínica apropiada y mediante pruebas de laboratorio. Si los niveles plasmáticos de actividad del factor VIII esperados no se alcanzan o si el sangrado no se controla con una dosis apropiada, se debe llevar a cabo la prueba para la detección de inhibidores del factor VIII. En pacientes con alto nivel de inhibidores, la terapia con factor VIII puede no ser efectiva y se deben considerar otras opciones terapéuticas. El manejo de estos pacientes debe ser dirigido por médicos con experiencia en el cuidado de individuos con hemofilia e inhibidores de factor VIII. Efectos cardiovasculares: En pacientes con factores de riesgo cardiovascular existentes, la terapia de sustitución con FVIII puede incrementar el riesgo cardiovascular. Complicaciones relacionadas con el catéter: Si se requiere un dispositivo de acceso venoso central (DAVC), se debe tener en cuenta el riesgo de complicaciones relacionadas con el DAVC, incluyendo infecciones locales, bacteriemia y trombosis en el lugar de inserción del catéter. Se recomienda fuertemente registrar el nombre y el número de lote del medicamento cada vez que se administre NovoEight® a un paciente con el fin de mantener un vínculo entre el paciente y el lote del medicamento. Población pediátrica: Las advertencias y precauciones indicadas son aplicables a adultos y niños. Consideraciones relacionadas con excipientes: Este medicamento contiene 30,5 mg de sodio por vial reconstituido, equivalente a 1,5% de la ingesta máxima diaria de 2 g de sodio recomendada por la OMS para un adulto. Interacción con otros medicamentos y otras formas de interacción: No se han reportado interacciones del factor VIII de coagulación humano (ADNr) con otros medicamentos. Embarazo, lactancia y fertilidad No se han llevado a cabo estudios de reproducción en animales con NovoEight®. Basados en la rara ocurrencia de hemofilia A en mujeres, no se dispone de experiencia con respecto al uso de factor VIII durante el embarazo y la lactancia. Por lo tanto, el factor VIII debe usarse durante el embarazo y lactancia solo si está claramente indicado. Efectos sobre la capacidad de conducir y operar máquinas: NovoEight® no tiene influencia en la capacidad de conducir o usar máquinas.
Incompatibilidades.
En ausencia de estudios de compatibilidad, este producto no debe ser mezclado con otros productos medicamentosos.
Conservación.
Almacenar en heladera (2°C - 8°C). No congelar. Mantener el vial dentro de su caja con la finalidad de protegerlo de la luz. La fecha de vencimiento está indicada en las etiquetas y en el estuche después de "Vence". Durante la vida útil, el producto puede ser almacenado a: temperatura ambiente (≤ 30°C) por un solo período no superior a 9 meses o por encima de la temperatura ambiente (de 30°C a 40°C) por un solo período no superior a 3 meses. Una vez que el producto ha sido retirado de la heladera, no se debe volver a refrigerar. Por favor, registre la fecha de inicio del almacenamiento y la temperatura de almacenamiento en el estuche del producto. Después de la reconstitución: Se ha demostrado la estabilidad física y química en uso por: 24 horas almacenado entre 2°C y 8°C. 4 horas almacenado a 30°C, para el producto que se haya conservado durante un único período no superior a 9 meses a temperatura ambiente (≤ 30°C). 4 horas almacenado a hasta 40°C, para el producto que se haya conservado durante un único período no superior a 3 meses por encima de la temperatura ambiente (de 30°C a 40°C). Desde el punto de vista microbiológico, el medicamento debería ser usado inmediatamente después de la reconstitución. Si el producto reconstituido no es utilizado inmediatamente, los tiempos y las condiciones de almacenamiento en uso antes de utilizarlo son responsabilidad del usuario y no deberían superar las arriba descritas, a menos que la reconstitución se haya realizado en condiciones asépticas controladas y validadas. Cualquier producto reconstituido almacenado a temperatura ambiente (≤ 30°C) o hasta 40°C que no se haya usado después de 4 horas debe ser desechado.
Sobredosificación.
No se han reportado síntomas por sobredosificación con el Factor VIII recombinante humano NovoEight®. Ante la eventualidad de una sobredosificación, concurrir al hospital más cercano o comunicarse con los centros de toxicología: Hospital de Pediatría Ricardo Gutiérrez: (011) 4962-6666/2247. Hospital A. Posadas: (011) 4654-6648/4658-7777. Opcionalmente, otros centros de intoxicaciones.
Presentación.
Cada envase de NovoEight® 250 UI, 500 UI, 1000 UI, 1500 UI, 2000 UI y 3000 UI polvo liofilizado y solvente para solución inyectable contiene: 1 vial de vidrio (Tipo I) con polvo y un tapón de goma de clorobutilo. 1 adaptador de vial estéril para reconstituir. 1 jeringa prellenada con 4 ml de solvente con un tope (polipropileno), un émbolo de goma (bromobutilo) y una tapa de jeringa con tapón (bromobutilo). 1 varilla del émbolo (polipropileno). Puede que no todas las presentaciones se encuentren comercializadas.
Revisión.
STF-Apr-2020_8-9063-00-006-1 Versión local 6.0.