ADCETRIS®
TAKEDA ARG.
Agente antineoplásico. Anticuerpo conjugado dirigido contra CD30.
Venta bajo receta archivada.
Composición.
Cada vial de Polvo Concentrado Para Reconstitución en Solución contiene: Brentuximab Vedotin 50 mg, Excipientes cs. Después de la reconstitución, cada ml contiene 5 mg de Brentuximab Vedotin.
Indicaciones.
Adcetris® está indicado para el tratamiento de pacientes con Linfoma de Hodgkin (LH) en recaída o refractario. Adcetris® está indicado para el tratamiento de pacientes con linfoma anaplásico de células grandes sistémico (LACGs) en recaída o refractario. Adcetris® está indicado para el tratamiento de pacientes con Linfoma de Hodgkin (LH) con riesgo de recaída o progresión seguido del TACM.
Dosificación.
Linfoma de Hodgkin y linfoma anaplásico de células grandes sistémico: Dosis: La dosis recomendada es 1,8 mg/kg administrados en forma de infusión intravenosa a lo largo de 30 minutos cada 3 semanas. Para pacientes con LH con riesgo de recaída o progresión después del TACM, Adcetris® se debe iniciar seguido de la recuperación al TACM basado en el juicio médico. Estos pacientes deben recibir hasta 16 ciclos. La dosis inicial recomendada para el tratamiento de pacientes con LH en recaída o refractario o LACGs quienes han respondido previamente al tratamiento con Adcetris® es 1,8 mg/kg administrada como una infusión intravenosa durante 30 minutos cada 3 semanas. Como alternativa, el tratamiento puede iniciarse con la última dosis tolerada. No debe administrarse en forma de inyección intravenosa rápida o en bolo. Si el paciente pesa más de 100 kg, la dosis debe calcularse usando un peso de 100 kg. Continuar el tratamiento hasta que haya progresión de la enfermedad o toxicidad inaceptable (véase Precauciones). Los pacientes con LH o LACGs en recaída o refractario que logren enfermedad estable o mejoría deben recibir un mínimo de 8 ciclos. Se tiene la experiencia clínica de tratar a pacientes hasta 16 ciclos (aproximadamente 1 año). En pacientes con insuficiencia renal severa la dosis recomendada de inicio es de1,2 mg/kg administrados en forma de infusión intravenosa a lo largo de 30 minutos cada 3 semanas. Los pacientes con insuficiencia renal deben ser estrechamente monitoreados para los eventos adversos. En pacientes con alteración de la función Hepática la dosis de inicio recomendada es de 1,2 mg/kg administrados en forma de infusión intravenosa a lo largo de 30 minutos cada 3 semanas. Los pacientes con insuficiencia hepática deben ser estrechamente monitoreados para los eventos adversos. Ajustes a la Dosis y/o Discontinuación de la Dosis: Modificación de la Dosis y/o Discontinuación de la Dosis: Continuar el tratamiento mientras el paciente continúe beneficiándose de y tolere el tratamiento.Véanse las recomendaciones para neuropatía periférica y neutropenia a continuación. Neuropatía Periférica: Si durante el tratamiento aparece o empeora una neuropatía periférica sensorial o motora, véanse las recomendaciones apropiadas en la Tabla 1 que se encuentra a continuación.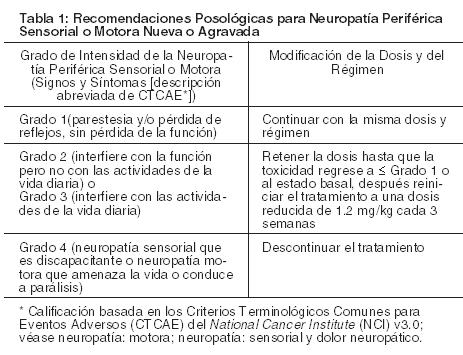
Neutropenia: Si se desarrolla neutropenia durante el tratamiento, ésta debe manejarse aplazando la dosis (véase Advertencias y Precauciones). Véanse las recomendaciones posológicas apropiadas en la Tabla 2 que se encuentra a continuación.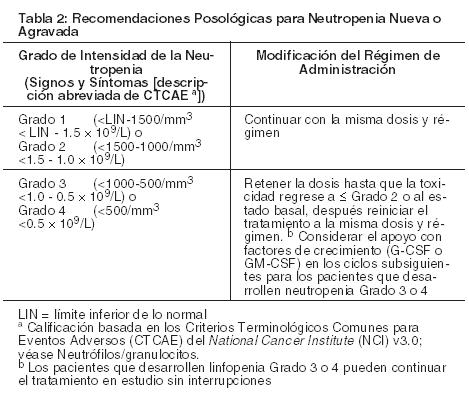
Contraindicaciones.
Hipersensibilidad al principio activo o a alguno de los excipientes. El uso en combinación de bleomicina y Adcetris® debido a que causa toxicidad pulmonar.
Reacciones adversas.
Experiencia de los estudios clínicos: Adcetris fue estudiado como monoterapia en 160 pacientes en 2 estudios pivotales de Fase 2 (SG035-0003 y SG035-0004) en pacientes con LH o LACGs en recaída o refractario. La media del número de ciclos fue 9 en pacientes con LH en recaída o refractario y 7 en los pacientes con LACGs en recaída o refractario. Adcetris también fue administrado como monoterapia en 167 de 329 pacientes en un estudio de Fase 3 aleatorizado controlado con placebo (SGN35-005) en pacientes con LH en riesgo de recaída o progresión tras TACM. La media del número de ciclos recibidos en ambos grupos fue de 15. La repetición del tratamiento con Adcetris se administró en 21 pacientes con LH en recaída o refractario y en 8 pacientes con LACGs en recaída (SGN 35-006). La media del número de ciclos fue de 7 (rango de 2 a 37 ciclos) (ver Estudios Clínicos). Los efectos adversos observados con mayor frecuencia (≥ 20%) en la población en los estudios pivotales de Fase 2 y el estudio de Fase 3 fueron neuropatía periférica, fatiga, náuseas, diarrea, infección del tracto respiratorio superior y neutropenia. Además, las reacciones adversas observadas también en ≥ 20% fueron vómitos y fiebre en los estudios pivotales de Fase 2 y neuropatía periférica motora y tos en el estudio de Fase 3. Lista tabulada de reacciones adversas: Las reacciones adversas para ADCETRIS se enumeran por el Sistema de clasificación de órganos MedDRA y término preferente (véase la Tabla 3) Dentro de cada clase de órganos y sistemas, las reacciones adversas se enumeran por categorías de frecuencia de: Muy frecuentes (≥ 1/10), frecuentes (≥ 1/100 a <1/10), poco frecuentes (≥ 1/1, 000, < 1/100).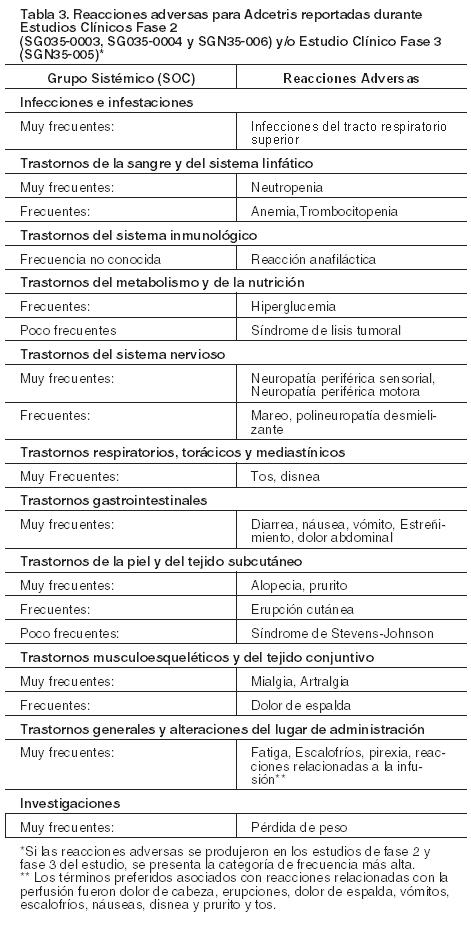
Reacciones adversas graves: Las reacciones adversas graves reportadas incluyen: neumonía, síndrome de distrés respiratorio agudo, dolor de cabeza, neutropenia, trombocitopenia, constipación, diarrea, vómitos, náuseas, pirexia, neuropatía motora periférica, neuropatía sensorial periférica, polineuropatía desmielinizante, síndrome de lisis tumoral y el Síndrome de Stevens-Johnson. Han sido reportados casos de Síndrome de Stevens-Johnson y necrólisis epidérmica tóxica en pacientes tratados con Adcetris (ver Precauciones). Han sido reportados casos de infecciones serias e infecciones oportunistas en pacientes tratados con Adcetris (ver Precauciones) Han sido reportados casos de pancreatitis aguda (incluyendo casos fatales) en pacientes tratados con Adcetris. Debe considerarse el diagnóstico de pancreatitis aguda en pacientes con dolor abdominal de reciente aparición o empeoramiento del mismo. Reacciones relacionadas con la infusión: En los estudios de Fase 2, fueron reportadas reacciones relacionadas con la infusión en 17 pacientes (11%) y en el 15% de los pacientes en el estudio Fase 3. Los eventos adversos más frecuentemente asociados a reacciones relacionadas con la infusión fueron de intensidad leve a moderada (Grado 1 o Grado 2) e incluyeron dolor de cabeza, erupciones, dolor de espalda, vómitos, escalofríos, náuseas, disnea, prurito y tos. Casos de anafilaxia reportados por fuera del contexto de los estudios clínicos de Fase 2 y Fase 3 que se están analizando en esta sección. Hepatotoxicidad: Fueron reportados casos de hepatotoxicidad manifestados por elevaciones asintomáticas de ALT y AST, estas elevaciones fueron leves a moderadas y transitorias en pacientes que reciben Adcetris. Toxicidad pulmonar: En los estudios clínicos se identificó la toxicidad pulmonar de la combinación de brentuximab vedotin con bleomicina (ver Contraindicaciones) y como un riesgo identificado en el PGR. Se han reportado casos de toxicidad pulmonar en pacientes que recibieron brentuximab vedotin y que no recibieron bleomicina. A pesar de que no se ha encontrado una relación causal, no puede excluirse la posibilidad de toxicidad pulmonar con brentuximab vedotin. Discontinuaciones: Los eventos adversos acontecidos llevaron a discontinuación del tratamiento en 23% y el 32% de los pacientes en los estudios pivotales de Fase 2 y el estudio de Fase 3, respectivamente. Los eventos adversos que llevaron a discontinuar el tratamiento en 2 o más pacientes tanto en los estudios pivotales de Fase 2 o en el estudio de Fase 3 fueron la neuropatía sensorial periférica, la neuropatía motora periférica, parestesia, vómitos y síndrome de dificultad respiratoria aguda. Modificaciones de dosis: La neutropenia llevó a retrasos en la dosis en el 14% y el 22% de los pacientes en los estudios pivotales de Fase 2 y el estudio de Fase 3, respectivamente. La neuropatía sensorial periférica dio lugar a retrasos en la dosis en el 13% y el 16% de los pacientes en los estudios pivotales de Fase 2 y el estudio de Fase 3, respectivamente. Además, la neuropatía periférica motora y la infección del tracto respiratorio superior, ambos condujeron a retrasos de la dosis en el 6% de los pacientes en el estudio de Fase 3. La neuropatía sensorial periférica condujo a una reducción de la dosis en el 9% y el 22% de los pacientes en los estudios pivotales de Fase 2 y el estudio de Fase 3, respectivamente. Además, la neuropatía periférica motora también condujo a la reducción de dosis en el 6% de los pacientes en el estudio de Fase 3. El noventa por ciento (90%) y sesenta y ocho por ciento (68%) de los pacientes en los estudios pivotales de Fase 2 y el estudio de Fase 3, respectivamente, se mantuvieron en la dosis recomendada de 1,8 mg/kg durante el tratamiento. Inmunogenicidad: A los pacientes con recaída o LH refractario o LACGs en dos estudios de Fase 2 (Ver Reacciones Adversas) se les realizaron las pruebas de anticuerpos para brentuximab vedotin cada 3 semanas utilizando un inmunoensayo electroquimioluminiscente sensible. También se probaron los pacientes con LH en riesgo de recaída o progresión tras TACM en el estudio de Fase 3. Aproximadamente el 7% de los pacientes en estos estudios pivotales de Fase 2 y el 6% de los pacientes del grupo ADCETRIS del estudio Fase 3 desarrollaron anticuerpos persistentemente positivos. Hubo una mayor incidencia de reacciones relacionadas con la infusión observadas en pacientes con anticuerpos positivos persistentes a ADCETRIS en relación con pacientes que dieron positivo o negativo de forma transitoria. Dos pacientes en los estudios pivotales en Fase 2 y dos pacientes en el estudio de Fase 3 experimentaron reacciones adversas consistentes con las reacciones relacionadas con la infusión que dieron lugar a la interrupción del tratamiento.La presencia de anticuerpos contra el brentuximab vedotin no se correlacionó con una reducción clínicamente significativa en los niveles de brentuximab vedotin suero y no dio lugar a una disminución en la eficacia de brentuximab vedotin. Los resultados de los ensayos de inmunogenicidad son altamente dependientes de varios factores, incluyendo sensibilidad y especificidad del ensayo, metodología del ensayo, manipulación de la muestra, tiempo de recolección de la muestra, medicaciones concomitantes y enfermedad de base. Por estas razones sería engañoso comparar la incidencia de anticuerpos con ADCETRIS con la incidencia de anticuerpos con otros productos. Descripción de reacciones adversas seleccionadas: Han sido reportados casos de Leucoencefalopatía multifocal progresiva (ver Precauciones) Ha sido reportada neutropenia febril (ver Precauciones). Retratamiento: Los tipos y grados de reacciones adversas notificados en pacientes retratados con ADCETRIS fueron consistentes con los observados en la combinación de los ensayos pivotales de Fase II, con la excepción de la neuropatía periférica motora, cuya incidencia fue mayor (28% vs 9% en los ensayos pivotales de Fase II) y fue principalmente de grados 1 o 2.
Precauciones.
Leucoencefalopatía Multifocal Progresiva: En los pacientes tratados con ADCETRIS puede presentarse infección por el virus de John Cunningham (VJC), que resulta en leucoencefalopatía multifocal progresiva (LMP) y muerte. Se ha notificado LMP en pacientes que recibieron ADCETRIS después de haber recibido múltiples regímenes de quimioterapia previos. La LMP es una enfermedad desmielinizante rara del sistema nervioso central que resulta de la reactivación del VJC latente y con frecuencia es mortal. Los pacientes deben ser estrictamente monitorizados ante la aparición o el agravamiento de signos o síntomas neurológicos, cognitivos o conductuales que pueden sugerir LMP. La administración de ADCETRIS debe suspenderse en cualquier caso de sospecha de LMP. La evaluación sugerida de LMP incluye consulta neurológica, resonancia magnética del cerebro con gadolinio y análisis del líquido cefalorraquídeo para detectar ADN del VJC por reacción en cadena de polimerasa o una biopsia cerebral que determine la presencia del VJC. Una reacción en cadena de polimerasa del VJC negativo no excluye la LMP. La administración de ADCETRIS debe discontinuarse permanentemente si se confirma el diagnóstico de LMP. El médico debe estar particularmente alerta a síntomas que sugieran LMP que el paciente pudiera no notar (por ejemplo, síntomas cognitivos, neurológicos o psiquiátricos). Pancreatitis: Han sido observados casos de pancreatitis en pacientes tratados con Adcetris. Algunos han sido fatales. Los pacientes deberán ser cuidadosamente monitoreados para detectar dolor abdominal de reciente aparición o empeoramiento, lo cual puede ser sugestivo de pancreatitis aguda. La evaluación del paciente puede incluir examen físico, determinación de amilasa y lipasa sérica, estudio por imágenes abdominal, como ultrasonido u otras imágenes. Ante la sospecha de pancreatitis, debe esperarse antes de administrar Brentuximab vedotin. Si se confirma el diagnóstico, deberá suspenderse el tratamiento definitivamente. Toxicidad pulmonar: Han sido reportado casos de toxicidad pulmonar, incluyendo neumonitis, enfermedad pulmonar intersticial y síndrome de distrés respiratorio agudo (ARDS), algunos con resultados fatales, en pacientes recibiendo Adcetris. A pesar de que no se ha determinado una relación causal, este efecto no puede ser descartado. En caso de aparición o empeoramiento de síntomas respiratorios (disnea, tos) deberá realizarse una evaluación diagnóstica inmediata y los pacientes deben ser tratados adecuadamente. Considere retener la posología de Adcetris durante la evaluación y hasta la mejoría sintomática. Infecciones Graves e Infecciones Oportunistas: Las infecciones graves y oportunistas como la neumonía, bacteriemia y sepsis/shock séptico (incluyendo desenlace fatal) han sido reportadas en pacientes tratados con brentuximab vedotin. Los pacientes deben ser monitorizados cuidadosamente durante el tratamiento por la aparición de posibles infecciones bacterianas, fúngicas o virales. Reacciones Relacionadas con la Infusión y reacciones de hipersensibilidad: Pueden ocurrir reacciones relacionadas con la infusión con Adcetris. Los pacientes deben ser monitorizados cuidadosamente durante y después de la infusión. Si se produce una reacción anafiláctica, la administración de brentuximab vedotin debe ser inmediata y permanentemente discontinuada y un apropiado tratamiento médico debe administrarse. Si se produce una reacción relacionada con la perfusión, la perfusión debe interrumpirse e instituirse un manejo médico adecuado. Los pacientes que hayan experimentado una reacción relacionada con la perfusión previa deben ser premedicados para infusiones posteriores. La premedicación puede incluir paracetamol (acetaminofén), un antihistamínico y un corticosteroide. Existen datos limitados con repetición del tratamiento de los pacientes que han sufrido una reacción anafiláctica con Adcetris. Neuropatía Periférica: El tratamiento con brentuximab vedotin puede causar neuropatía periférica, tanto sensorial y motora. La neuropatía periférica inducida por brentuximab vedotin es típicamente un efecto acumulativo y es reversible en la mayoría de los casos. En la población de Fase 2, en el momento de la última evaluación, la mayoría de los pacientes (62%) tuvieron una mejoría o la resolución de sus síntomas de neuropatía periférica. Para los pacientes que informaron de la neuropatía periférica, la discontinuación del tratamiento con brentuximab vedotin se produjo en un 9%, se registraron reducciones de dosis en el 8%, y retrasos de dosis en el 13% de los pacientes. Los pacientes deben ser monitorizados para detectar síntomas de la neuropatía, como hipoestesia, hiperestesia, parestesia, malestar, una sensación de ardor, dolor neuropático o debilidad. Los pacientes que presentaron nuevos casos o agravamiento de la neuropatía periférica pueden requerir un retraso y una reducción de la dosis de brentuximab vedotin o suspensión del tratamiento. Toxicidad Hematológica: Anemia grado 3 o grado 4, trombocitopenia y neutropenia prolongada (≥ 1 semana) puede ocurrir con brentuximab vedotin. Se reportó neutropenia febril con el tratamiento con Adcetris. Deben realizarse hemogramas completos de control antes de la administración de cada dosis. Los pacientes deben ser monitoreados de cerca para fiebre. Si se desarrolla neutropenia de grado 3 o grado 4, manejar con modificaciones de dosis o interrupción. Síndrome de Lisis Tumoral: Se ha notificado síndrome de lisis tumoral con ADCETRIS. Los pacientes con tumor en rápida proliferación y elevada carga tumoral están en riesgo de padecer síndrome de lisis tumoral. Estos pacientes deben mantenerse bajo estrecha observación y se deben tomar las medidas apropiadas. Síndrome de Stevens-Johnson: Se ha notificado síndrome de Stevens-Johnson (SSJ) y necrólisis epidérmica tóxica (NET) con ADCETRIS. Se han notificado casos mortales. Si se presenta el síndrome de Stevens-Johnson o NET, se debe discontinuar ADCETRIS y administrarse el tratamiento médico apropiado. Complicaciones Gastrointestinales: Fueron reportadas complicaciones gastrointestinales (GI), en pacientes tratados con Adcetris, incluyendo obstrucción intestinal, íleo, enterocolitis, colitis neutropénica, erosión, úlcera, perforación y hemorragia, algunas con resultados fatales. Se reportaron algunos casos de perforaciones gastrointestinales en pacientes con afección gastrointestinal de un linfoma subyacente. En caso de síntomas GI nuevos o empeoramiento, realizar una rápida evaluación de diagnóstico y tratar de forma adecuada. Hepatotoxicidad: Se reportó hepatotoxicidad en forma de elevaciones de la alanina aminotransferasa (ALT) y aspartato aminotransferasa (AST) con Adcetris. También ocurrieron casos graves de hepatotoxicidad, incluyendo resultados fatales. Una enfermedad hepática pre-existente, comorbilidades y medicaciones concomitantes también pueden aumentar el riesgo. Debe monitorearse la función hepática de forma rutinaria en pacientes que reciben brentuximab vedotin. Los pacientes que experimentan hepatotoxicidad pueden requerir un retraso, cambio en la dosis o la interrupción de Adcetris (Ver Reacciones adversas) Hiperglucemia: Hiperglucemia ha sido reportada en los estudios clínicos en pacientes con un índice de masa corporal elevado, con o sin antecedentes de diabetes. Si un paciente presenta un evento de hiperglucemia, deberá monitorearse la glucemia de cerca. Deberá instaurarse un tratamiento antidiabético si es necesario. Contenido de sodio en excipientes: Este medicamento contiene un máximo de sodio de 2,1 mmol (ó 47 mg) de sodio por dosis. Esto deberá tenerse en cuenta en pacientes con dietas pobres en sodio.
Presentación.
Vial x 1.