REVIXIL® 20
GADOR
Movilizador de células madre hematopoyéticas; otros inmunoestimulantes.
Composición.
Cada mililitro de solución inyectable de REVIXIL® 20 contiene: Plerixafor 20 mg. Excipientes: Cloruro de sodio 4,917 mg, Ácido clorhídrico concentrado 0,013 ml, Agua para inyección c.s.p. 1 ml.
Farmacología.
El mecanismo de acción de plerixafor es la inhibición del receptor de quimiocina CXCR4, bloqueando de esta forma la unión del ligando análogo, el factor-1a derivado de las células estromales (SDF-1a). Este receptor y su ligando cumplen una importante función en la circulación y alojamiento de las células madre hematopoyéticas en el compartimiento medular. El CXCR4 puede ayudar a fijar las células madre a la matriz medular, ya sea a través del ligando SDF-1a o mediante la inducción de otras moléculas de adhesión. Plerixafor produce leucocitosis y aumento de las células madre hematopoyéticas circulantes, resultado de una alteración de la unión de CXCR4 con su ligando afín, lo que da lugar a la aparición tanto de células maduras como pluripotentes en la circulación sistémica. Las células CD34+ movilizadas por plerixafor son funcionales y pueden injertarse con capacidad de repoblación a largo plazo. El pico de movilización de células CD34+, que se ha demostrado en estudios de farmacodinamia con plerixafor, ocurre entre las 6 y 9 horas luego de su administración. Este pico se observa entre las 10 y 14 hs en la administración conjunta de plerixafor con G-CSF.
Farmacocinética.
Se han evaluado los parámetros farmacocinéticos de plerixafor en pacientes con linfoma y mieloma múltiple al nivel de dosis clínica de 0,24 mg/kg después del pretratamiento con G-CSF (10 mg/kg una vez al día durante 4 días consecutivos). Absorción: Luego de la administración de una dosis subcutánea, las concentraciones plasmáticas máximas de plerixafor se consiguen a los 30-60 minutos. Tras la administración subcutánea de una dosis de 0,24 mg/kg a los pacientes después de recibir 4 días de pretratamiento con G-CSF, la concentración plasmática máxima (Cmáx) y la exposición sistémica (ABC0-24) de plerixafor eran de 887 ± 217 ng/ ml y 4.337 ± 922 ng.h/ml, respectivamente. Distribución: Plerixafor se une moderadamente a las proteínas plasmáticas humanas, hasta en un 58%. El volumen aparente de distribución de plerixafor en humanos es de 0,3 l/kg, lo que demuestra que plerixafor está confinado en gran medida, aunque no exclusivamente, al espacio extracelular líquido. Metabolismo: Plerixafor no se metaboliza in vitro cuando se usan microsomas hepáticos humanos o hepatocitos primarios humanos y no presenta actividad inhibitoria in vitro frente a las principales enzimas del citocromo P450 que metabolizan fármacos (1A2, 2A6, 2B6, 2C8, 2C9, 2C19, 2D6, 2E1 y 3A4/5). En estudios in vitro con hepatocitos humanos, plerixafor no induce las enzimas del citocromo P1A2, del citocromo P2B6 ni del citocromo P3A4. Estos resultados sugieren que existe una baja posibilidad de que plerixafor esté implicado en interacciones entre medicamentos dependientes del citocromo P450. Eliminación: La ruta principal de eliminación de plerixafor es la urinaria. Tras una dosis de 0,24 mg/kg en voluntarios sanos con función renal normal, aproximadamente el 70% de la dosis se excretaba sin metabolizar en la orina durante las primeras 24 horas tras su administración. La semivida de eliminación (t1/2) en plasma es de 3-5 horas. Plerixafor no actuó como sustrato o inhibidor de la glicoproteína P en un estudio in vitro con modelos celulares MDCKII y MDCKII-MDR1. Poblaciones especiales: Insuficiencia renal: Tras la administración de una única dosis de 0,24 mg/kg de plerixafor, el aclaramiento se reducía en los pacientes con grados variables de insuficiencia renal y se correlacionaba positivamente con el aclaramiento de creatinina (CrCl). Los valores medios de ABC0-24 de plerixafor en sujetos con insuficiencia renal leve (CrCl, 51-80 ml/ min), moderada (CrCl, 31-50 ml/min) y grave (CrCl, -30 ml/min) eran 5410, 6780 y 6990 ng.h/ml, respectivamente, que eran superiores a los observados en sujetos sanos con una función renal normal (5070 ng h/ml). La insuficiencia renal no afectaba a la Cmáx. Sexo: Un análisis farmacocinético poblacional evidenció que el género no produce efectos sobre la farmacocinética del plerixafor. Pacientes de edad avanzada: Un análisis farmacocinético poblacional evidenció que la edad no produce efectos sobre la farmacocinética del plerixafor. Población pediátrica: En base a modelos farmacocinéticos poblacionales y similares a los adultos, la dosificación en mg/kg resulta en un aumento en la exposición a plerixafor con incremento del peso corporal en pacientes pediátricos. En las mismas pautas de dosificación basadas en el peso de 240 mg/kg, la exposición media a plerixafor (ABC0-24h) es menor en pacientes pediátricos de 2 a < 6 años (1.410 ng.h/mL), de 6 a < 12 años (2.318 ng.h/mL), y de 12 a < 18 años (2.981 ng.h mL) que en adultos (4.337 ng.h/mL). En base al modelo farmacocinético poblacional, las exposiciones medias a plerixafor (ABC0-24h) en pacientes pediátricos de 2 a < 6 años (1.905 ng.h mL), de 6 a < 12 años (3.063 ng.h/mL) y de 12 a < 18 años (4.015 ng.h/mL), a la dosis de 320 mg/kg están más cerca de la exposición en adultos que reciben 240 mg/kg.
Indicaciones.
Pacientes adultos: REVIXIL® 20 está indicado, en combinación con el factor estimulante de granulocitos (G-CSF), para potenciar la movilización de las células madre hematopoyéticas a sangre periférica para su recolección y posterior trasplante autólogo, en pacientes adultos con linfoma no Hodgkin y mieloma múltiple. Pacientes pediátricos (de 1 año a menos de 18 años): REVIXIL® 20 está indicado en combinación con G-CSF para potenciar la movilización de células madre hematopoyéticas a sangre periférica para su recolección y posterior trasplante autólogo en niños con linfoma o tumores sólidos malignos, ya sea: - de forma preventiva, cuando se considera que el recuento de células madre circulantes en el día previsto de recogida, después de la movilización adecuada con G-CSF (con o sin quimioterapia), es insuficiente respecto al rendimiento deseado de células madre hematopoyéticas, o - cuando no se logra recolectar de forma previa suficientes células madre hematopoyéticas.
Dosificación.
El tratamiento con REVIXIL® 20 debe ser iniciado y supervisado por un médico especialista en oncología y/o hematología. Los procedimientos de movilización y aféresis deben realizarse en colaboración con un centro de oncología-hematología con experiencia apropiada en este campo y en el que se pueda realizar correctamente el control de las células madre hematopoyéticas. Se han identificado como indicadores de una movilización escasa tener más de 60 años y/o la quimioterapia previa mielosupresora y/o la quimioterapia previa extensiva y/o un pico en el número de células madre circulantes de menos de 20 células madre/microlitro. Posología: Pacientes adultos: La dosis diaria recomendada de REVIXIL® 20 es: Dosis fija de 20 mg o 0,24 mg/kg de peso corporal, en pacientes que pesan 83 kg o menos. 0,24 mg/kg de peso corporal, en pacientes que pesan más de 83 kg. Pacientes pediátricos (de 1 año a menos de 18 años): La dosis diaria recomendada de REVIXIL® 20 es: • 0,24 mg/kg de peso corporal. Para calcular la dosis de plerixafor se debe utilizar el peso del paciente medido en la semana previa a la primera administración de plerixafor. En los estudios clínicos, la dosis de plerixafor se ha calculado basándose en el peso de pacientes con un porcentaje de hasta el 175% superior a su peso ideal. No se ha estudiado la dosis de plerixafor y el tratamiento de pacientes con un peso superior al 175% de su peso ideal. El peso ideal se puede determinar usando las siguientes ecuaciones: - hombres (kg): 50 + 2,3 x [(estatura en cm x 0,394) - 60]; - mujeres (kg): 45,5 + 2,3 x [(estatura en cm x 0,394) - 60]. Cada frasco ampolla de REVIXIL® 20 contiene 1,2 mL de solución con 20 mg de plerixafor/mL (24 mg de plerixafor); por lo tanto el volumen a administrar surge de la siguiente fórmula: 0,012 x peso corporal actual (en kg) del paciente = volumen a administrar (en mL). En función del aumento de exposición con el aumento del peso corporal, la dosis de plerixafor no debe superar los 40 mg/día. REVIXIL® 20 se debe preparar en un tamaño de jeringa que se debe seleccionar de acuerdo con el peso del paciente. Para pacientes de peso bajo, hasta 45 kg de peso corporal, se pueden utilizar jeringas de uso infantil de 1 ml. Este tipo de jeringa tiene graduaciones mayores de 0,1 ml y graduaciones menores de 0,01 ml y, por lo tanto, es adecuado para administrar plerixafor, en una dosis de 240 mg/ kg, a pacientes pediátricos de al menos 9 kg de peso corporal. Para pacientes de más de 45 kg, se puede utilizar una jeringa de 1 ml o 2 ml con graduaciones que permitan medir un volumen de 0,1 ml. REVIXIL® 20 se administra por vía subcutánea de 6 a 11 horas antes del inicio de cada aféresis y después de pretratamiento con G-CSF de 4 días de duración. Plerixafor se emplea comúnmente durante 2 a 4 días consecutivos, y se ha utilizado por hasta 7 días consecutivos en el contexto de estudios clínicos. Medicamentos concomitantes: El G-CSF se administra en dosis matutinas de 10 microgramos/ kg en los 4 días previos al comienzo de la dosis vespertina de REVIXIL® 20 y cada mañana antes de la aféresis. Poblaciones especiales: Insuficiencia renal: En los pacientes con CrCl de 20 a 50 ml/min se deberá reducir la dosis de REVIXIL® 20 en un tercio hasta 0,16 mg/kg/día, de acuerdo a la categoría establecida en función del peso corporal, como se muestra en la Tabla 1. Los datos clínicos que evalúan este ajuste de dosis son limitados. No se cuenta con suficiente experiencia clínica para hacer recomendaciones posológicas alternativas en pacientes con un CrCl < 20 ml/min o en hemodiálisis. La dosis no debe exceder los 27 mg/día si el CrCl es -50 mL/min, en base al aumento en la exposición con el incremento del peso corporal.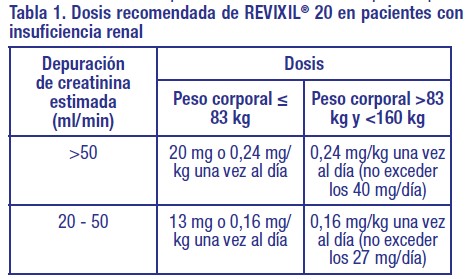
Pacientes de edad avanzada ( > 65 años): No es necesario realizar ningún ajuste de la dosis en pacientes de edad avanzada con función renal normal. Sin embargo, sí se recomienda realizar un ajuste de dosis en pacientes de edad avanzada con un CrCl -50 ml/min (ver arriba el apartado Insuficiencia renal). En general, hay que tener precaución en la elección de la dosis de pacientes de edad avanzada, debido a la mayor frecuencia de disminución de la función renal en pacientes con edad avanzada. Forma de administración: REVIXIL® 20 es para inyección subcutánea. Cada frasco ampolla está destinado para un uso único, debiendo desecharse el resto del fármaco no utilizado. Antes de su administración, cada frasco ampolla de REVIXIL® 20 deberá ser inspeccionado visualmente y no deberá utilizarse en caso de presentar material particulado en suspensión o un cambio de color. Debe emplearse una técnica aséptica cuando se transfiera el contenido del frasco ampolla a una jeringa adecuada para su administración subcutánea.
Contraindicaciones.
Hipersensibilidad conocida al principio activo o a los componentes de REVIXIL® 20.
Reacciones adversas.
A continuación se enumeran las reacciones adversas aparecidas con el uso de plerixafor, tanto en estudios clínicos como durante la experiencia poscomercialización. Las mismas se agrupan según el sistema de clasificación de órganos y por frecuencia. Las frecuencias se definen de la siguiente forma: frecuentes (mayor del 10%); ocasionales (1% al 10%); raras (menor del 1%); frecuencia no conocida (no puede estimarse a partir de los datos disponibles). Las reacciones adversas se presentan en orden decreciente de gravedad dentro de cada intervalo de frecuencia. Debido a que los estudios clínicos se conducen bajo condiciones ampliamente variables, las tasas de eventos adversos observados en los estudios clínicos de un fármaco no son directamente comparables con las tasas de los estudios clínicos de otro fármaco, y pueden no reflejar las tasas observadas en la práctica. Asimismo, las reacciones adversas identificadas luego de la aprobación del medicamento son reportadas voluntariamente a partir de una población de tamaño desconocido, por lo que no siempre es posible estimar en forma confiable la frecuencia o establecer una relación causal con la exposición al fármaco. Trastornos de la sangre y del sistema linfático. Ocasionales: hiperleucocitosis. Frecuencia no determinada: plaquetopenia, esplenomegalia, rotura esplénica. Trastornos del sistema inmunológico. Raras: reacciones alérgicas (incluyendo urticaria, inflamación periorbital, disnea o hipoxia) -efectos generalmente leves o moderados, apareciendo aproximadamente en los 30 minutos siguientes a la administración de plerixafor-, reacciones anafilácticas (incluyendo shock anafiláctico). Trastornos psiquiátricos. Ocasionales: insomnio. Raras: sueños anormales, pesadillas. Trastornos del sistema nervioso. Ocasionales: mareos, cefaleas. Trastornos gastrointestinales. Frecuentes: diarrea, náuseas. Ocasionales: vómitos, dolor abdominal, molestias estomacales, dispepsia, distensión abdominal, estreñimiento, flatulencia, hipoestesia oral, sequedad en la boca. Trastornos de la piel y del tejido subcutáneo. Ocasionales: hiperhidrosis, eritema. Trastornos musculoesqueléticos y del tejido conjuntivo. Ocasionales: artralgias, dolor musculoesquelético. Trastornos generales y alteraciones en el lugar de administración. Frecuentes: reacciones en el sitio de inyección (incluyendo eritema, hematoma, hemorragia, induración, inflamación, irritación, dolor, parestesia, prurito, erupción, hinchazón y urticaria). Ocasionales: fatiga, malestar. No se observaron diferencias significativas en la incidencia de reacciones adversas en los pacientes oncológicos por enfermedad, sexo o edad. Descripción de reacciones adversas seleccionadas: Infarto de miocardio: En estudios clínicos con plerixafor, 7 de los 679 pacientes oncológicos sufrieron infartos de miocardio después de la movilización de células madre hematopoyéticas con plerixafor y G-CSF. Todos los acontecimientos se produjeron al menos 14 días después de la última administración de plerixafor. Además, en el programa de uso compasivo, dos pacientes oncológicos de sexo femenino sufrieron infarto de miocardio tras la movilización de células madres hematopoyéticas con plerixafor y G-CSF. Una de estas reacciones se produjo 4 días después de la última administración de plerixafor. La falta de relación temporal en 8 de los 9 pacientes junto con el perfil de riesgo de los pacientes con infarto de miocardio, no sugiere que plerixafor produzca un riesgo independiente de infarto de miocardio en pacientes que también reciben G-CSF. Hiperleucocitosis: En los estudios en fase III con plerixafor se observaron recuentos de leucocitos de 100 x 109/L o superior, el día anterior o cualquiera de los días de aféresis, en el 7% de los pacientes que recibieron plerixafor y en el 1% de los pacientes que recibieron placebo. No se observaron complicaciones ni síntomas clínicos de leucostasis. Reacciones vasovagales: En estudios clínicos con plerixafor en pacientes oncológicos y voluntarios sanos, menos del 1% de los sujetos experimentaron reacciones vasovagales (hipotensión ortostática y/o síncope) tras la administración subcutánea de la dosis de plerixafor -0,24 mg/kg. La mayoría de estas reacciones se produjeron en el lapso de 1 hora tras la administración de plerixafor. Trastornos gastrointestinales: En los estudios clínicos con plerixafor en pacientes oncológicos, se han descrito en raras ocasiones reacciones gastrointestinales severas, como diarrea, náuseas, vómitos y dolor abdominal. Parestesias: La parestesia se observa frecuentemente en pacientes oncológicos sometidos a trasplante autólogo tras intervenciones múltiples relacionadas con la enfermedad. En los estudios en fase III controlados con placebo, la incidencia de parestesia fue del 20,6% y del 21,2% en los grupos de plerixafor y placebo, respectivamente. Pacientes de edad avanzada: En los dos estudios clínicos controlados con placebo de plerixafor, el 24% de los pacientes tenía 65 años o más. No se observaron diferencias destacables en la incidencia de reacciones adversas en estos pacientes de edad avanzada cuando se comparaban con pacientes más jóvenes. Pacientes pediátricos: El perfil de seguridad de plerixafor en un estudio clínico con pacientes pediátricos fue concordante con el observado en adultos. Notificación de Sospecha de Reacciones Adversas Es importante notificar la sospecha de reacciones adversas al medicamento tras su autorización. Ello permite una supervisión continuada de la relación beneficio/riesgo del medicamento. Se invita a los profesionales sanitarios a notificar las sospechas de reacciones adversas asociadas al uso de REVIXIL® 20 a través del Sistema Nacional de Farmacovigilancia al siguiente link: https:// www.argentina.gob.ar/anmat/farmacovigilancia/notificanos/eventosadversos y/o al Departamento de Farmacovigilancia de GADOR S.A. vía email a farmacovigilancia@gador.com o telefónicamente al 0800-220-2273.
Advertencias.
Shock anafiláctico y reacciones de hipersensibilidad: Se ha notificado la aparición de reacciones alérgicas de leve a moderada intensidad dentro de los 30 minutos de la administración de plerixafor, en menos del 1% de los pacientes tratados en estudios clínicos. Plerixafor podría asociarse con reacciones sistémicas relacionadas a la inyección subcutánea, como urticaria, edema periorbitario, disnea o hipoxia; los síntomas han respondido al tratamiento (por ejemplo, antihistamínicos, corticoesteroides, hidratación u oxígeno suplementario) o resolvieron espontáneamente. Se ha informado la ocurrencia de reacciones de hipersensibilidad graves, incluyendo aquellas de tipo anafiláctico con riesgo de muerte e hipotensión clínicamente significativa y shock, en pacientes que recibían plerixafor. Se debe controlar a los pacientes para detectar la aparición de signos y síntomas de hipersensibilidad durante y luego de la administración de REVIXIL® 20, por al menos 30 minutos y hasta la estabilidad clínica, posteriormente a cada administración. REVIXIL® 20 solo debe ser administrado cuando se disponga en forma inmediata de personal y recursos terapéuticos para el tratamiento de reacciones anafilácticas u otras reacciones de hipersensibilidad. Movilización de células tumorales en pacientes con linfoma y mieloma múltiple: La administración conjunta de REVIXIL® 20 con G-CSF puede producir la liberación de células tumorales desde la médula ósea y la subsecuente recolección en el producto de la aféresis. Los resultados mostraron que, en el caso de que las células tumorales se movilicen, el número de células tumorales movilizadas no aumenta con plerixafor más G-CSF en comparación con G-CSF solo. Movilización de células tumorales en pacientes con leucemia: El uso de REVIXIL® 20 para la movilización de células madre hematopoyéticas podría causar también la movilización de células leucémicas y la subsiguiente contaminación del producto de la aféresis. Por este motivo, REVIXIL® 20 no se recomienda para la movilización y recolección de células madre hematopoyéticas en pacientes con leucemia. Efectos hematológicos: Hiperleucocitosis. La administración de plerixafor junto con G-CSF aumenta el número de leucocitos circulantes, así como las poblaciones de células madre hematopoyéticas. Se deberá monitorear el recuento de glóbulos blancos durante el tratamiento con REVIXIL® 20. La administración de REVIXIL® 20 a los pacientes con un recuento de neutrófilos en sangre periférica superior a 50 x 109/L debe basarse en el criterio clínico. Trombocitopenia. La trombocitopenia es una complicación conocida de la aféresis y se ha observado en pacientes que recibieron plerixafor. Se deberá controlar el recuento plaquetario en los pacientes bajo tratamiento con REVIXIL® 20 que posteriormente sean sometidos a la aféresis. Esplenomegalia y posible ruptura esplénica: Se ha observado en ratas un aumento del peso del bazo junto a hematopoyesis extramedular, luego de la administración prolongada de plerixafor (hasta 4 semanas) con dosis aproximadamente 4 veces superiores a la recomendada en seres humanos. Este efecto del plerixafor no ha sido evaluado específicamente en estudios clínicos. Se han notificado casos de esplenomegalia y/o rotura esplénica tras la administración de plerixafor junto con el factor de crecimiento G-CSF. Se deberá controlar la integridad del bazo en pacientes que reciben tratamiento conjunto con REVIXIL® 20 y G-CSF y que presenten dolor en hipocondrio izquierdo y/o escapular o en los hombros. Reacciones vasovagales: Luego de la administración subcutánea de plerixafor, pueden ocurrir reacciones vasovagales, hipotensión ortostática y/o síncope, por lo que se deben tomar las precauciones apropiadas. Toxicidad embriofetal: No existen datos suficientes sobre la utilización de plerixafor en mujeres embarazadas. Su mecanismo de acción farmacodinámico sugiere que plerixafor puede causar malformaciones congénitas cuando se administra durante el embarazo. Los estudios en animales han mostrado teratogenicidad. Interacciones medicamentosas y otras formas de interacción No se han realizado estudios de interacciones. Plerixafor no es sustrato, inhibidor o inductor de las isoenzimas del citocromo P450 en estudios in vitro, por lo que es poco probable que ocurran interacciones farmacológicas in vivo dependientes de estas enzimas. Plerixafor no actuó como sustrato ni inhibidor de la glucoproteína-P en estudios in vitro. En los estudios clínicos en pacientes con linfoma no Hodgkin, la adición de rituximab al tratamiento de movilización con plerixafor y G-CSF no afectó a la seguridad de los pacientes ni al rendimiento de células CD34+. Incompatibilidades: En ausencia de estudios de compatibilidad, REVIXIL® 20 no se debe mezclar con otros medicamentos. Efectos sobre la capacidad para conducir o utilizar maquinarias: REVIXIL® 20 puede afectar la capacidad para conducir o utilizar maquinarias. Algunos pacientes han experimentado mareos, fatiga o reacciones vasovagales; se recomienda precaución al conducir u operar maquinarias. Carcinogénesis, mutagénesis, compromiso de la fertilidad, otros datos preclínicos sobre seguridad: Plerixafor no ha sido estudiado en carcinogenicidad. Plerixafor no presentó genotoxicidad en los estudios de Ames, de aberración cromosómica en células de ovario de hámster chino, ni en la prueba in vivo de micronúcleos de médula ósea en rata. No se han realizado estudios toxicológicos reproductivos. No se observaron signos de toxicidad en los órganos reproductores masculinos o femeninos, en los estudios de toxicidad de dosis repetida de 28 días en ratas. En estudios de distribución en ratas, se detectaron concentraciones de plerixafor marcado radioactivamente en órganos reproductores (testículos, ovarios y útero) dos semanas después de la administración de una dosis única o de 7 dosis repetidas diariamente en machos y de 7 dosis repetidas diariamente en hembras. La tasa de eliminación de los tejidos era lenta. No se conocen los efectos del plerixafor sobre la fertilidad masculina y femenina. SDF-1a y CXCR4 desempeñan funciones importantes en el desarrollo embriofetal. Se ha demostrado que plerixafor causa el aumento de reabsorciones, reducción de los pesos fetales, retraso del desarrollo esquelético y aumento de las anomalías fetales en ratas y conejos. Los datos de modelos animales también sugieren que SDF-1a y CXCR4 modulan la hematopoyesis fetal, la vascularización y el desarrollo del cerebelo. La exposición sistémica al nivel sin efectos adversos observables (NOAEL) para efectos teratogénicos en ratas y conejos fue de la misma magnitud o inferior a las dosis terapéuticas en pacientes. Este potencial teratogénico es debido probablemente a su mecanismo de acción farmacodinámico. No se han evaluado en estudios no clínicos los posibles efectos de plerixafor sobre el desarrollo postnatal. Los resultados de los estudios a dosis únicas subcutáneas en ratas y ratones mostraron que plerixafor puede inducir efectos neuromusculares transitorios pero graves (movimientos descoordinados), efectos similares a la sedación (hipoactividad), disnea, inactividad en posición de decúbito prono o lateral y/o espasmos musculares. Efectos adicionales de plerixafor observados sistemáticamente en estudios animales a dosis repetidas fueron: aumento de los niveles de leucocitos circulantes y de la excreción urinaria de calcio y magnesio en ratas y perros, pesos ligeramente superiores del bazo en ratas y diarrea y taquicardia en perros. Los hallazgos histopatológicos de hematopoyesis extramedular se observaron en hígado y bazo de ratas y/o perros. Uno o más de estos hallazgos se observaban normalmente con exposiciones sistémicas del mismo orden de magnitud o ligeramente superior que la exposición clínica en humanos. Los resultados del estudio de ajuste de dosis en cerdos enanos jóvenes y el ajuste de dosis y estudios definitivos en ratas jóvenes fueron similares a los observados en ratones adultos, ratas y perros. Los márgenes de exposición en el estudio con ratas jóvenes a la dosis máxima tolerada (DMT) fueron 18 veces mayores comparado con la dosis clínica pediátrica más alta en niños de hasta 18 años. Una prueba in vitro general de actividad de receptores mostró que plerixafor, a una concentración (5 mg/ml) varias veces superior al nivel sistémico humano, tiene una afinidad de unión fuerte o moderada con varios receptores ubicados principalmente en los extremos de nervios presinápticos en el sistema nervioso central (SNC) y/o el sistema nervioso periférico (SNP) (receptores del canal de calcio tipo N, canal SKCA de potasio, histamina H3, M1 y M2 de acetilcolina muscarínica, adrenérgico a1B y a2C, neuropéptido Y/Y1 y poliamina NMDA glutamato). Se desconoce la relevancia clínica de estos resultados. Los estudios de farmacología de seguridad con plerixafor administrado por vía intravenosa en ratas mostraron efectos depresores respiratorios y cardiacos con exposiciones sistémicas ligeramente por encima de la exposición clínica en humanos, mientras que la administración subcutánea solo provocó efectos respiratorios y cardiacos con exposiciones sistémicas mayores. Plerixafor inhibió el crecimiento tumoral en modelos in vivo de linfoma no Hodgkin, glioblastoma, meduloblastoma y leucemia linfoblástica aguda cuando las dosis se administraban de forma intermitente. Se observó un aumento del crecimiento del linfoma no Hodgkin después de la administración continua de plerixafor durante 28 días. Se espera que el riesgo potencial asociado con este efecto sea bajo para la administración de plerixafor en humanos proyectada a corto plazo. Mujeres en edad fértil: Se deberá recomendar a las mujeres en edad reproductiva evitar el embarazo durante el tratamiento con REVIXIL® 20 y hasta una semana luego de la dosis final, utilizando un método anticonceptivo efectivo. Realizar pruebas de embarazo en mujeres en edad fértil antes de comenzar el tratamiento con REVIXIL® 20. Embarazo: No hay estudios adecuados y controlados en mujeres embarazadas utilizando plerixafor; en base al mecanismo farmacodinámico de acción, se sugiere que plerixafor puede causar malformaciones congénitas si se administra durante el embarazo. Durante el embarazo, REVIXIL® 20 debe utilizarse sólo en caso de que el beneficio potencial para la madre justifique el riesgo potencial para el feto. Si REVIXIL® 20 se empleara durante el embarazo, o si la paciente quedara embarazada mientras utiliza REVIXIL® 20, se debe informar a la paciente de los riesgos potenciales para el feto. Lactancia: No se conoce aún si plerixafor se excreta por la leche humana. Dado que muchos medicamentos pueden excretarse por esta vía y debido al potencial riesgo de reacciones adversas serias a plerixafor en lactantes, debe interrumpirse la lactancia durante el tratamiento con REVIXIL® 20 y durante una semana luego de la dosis final. Empleo en geriatría: No se han observado diferencias en la seguridad y efectividad de la droga entre pacientes jóvenes y pacientes de 65 años o más. No es necesario modificar la dosis en adultos mayores con función renal normal, aunque se deberá proceder con cautela al seleccionar la dosis ya que en estos pacientes de edad avanzada puede observarse una disminución de la función renal. Se recomienda ajustar la dosis de REVIXIL® 20 cuando el CrCl sea -50 mL/min (ver Dosificación).
Conservación.
Conservar en su envase original a temperatura ambiente hasta 30°C.
Sobredosificación.
No se han notificado casos de sobredosis. Basándose en los datos limitados obtenidos con dosis por encima de la dosis recomendada y de hasta 0,48 mg/kg, la frecuencia de reacciones vasovagales, hipotensión ortostática y/o sincope y trastornos gastrointestinales puede aumentar. Ante la eventualidad de una sobredosificación, concurrir al Hospital más cercano o comunicarse con los Centros de Toxicología: Hospital de Pediatría Ricardo Gutiérrez: (011) 4962-6666/2247. Hospital Alejandro Posadas: (011) 4654-6648/4658-7777. Optativamente otros centros de intoxicaciones.
Presentación.
Envases con 1 frasco ampolla conteniendo 1,2 ml de solución inyectable.
Revisión.
11/2020. G00130801-02.