INLYTA 1MG
PFIZER
Agente antineoplásico.
Composición.
Cada comprimido de INLYTA 1 mg contiene: Axitinib 1,000 mg. Celulosa microcristalina 63,250 mg. Lactosa monohidrato 32,000 mg. Croscarmelosa sódica 3,000 mg. Estearato de magnesio 0,750 mg. Opadry II rojo 4,000 mg. Cada comprimido de INLYTA 5 mg contiene: Axitinib. 5,000 mg. Celulosa microcristalina 107,430 mg. Lactosa monohidrato 56,000 mg. Croscarmelosa sódica 5,250 mg. Estearato de magnesio 1,320 mg. Opadry II rojo 7,000 mg.
Farmacología.
Descripción: INLYTA (axitinib) es un inhibidor de quinasas. El nombre químico de axitinib es N-Metil-2-[3-((E)-2-piridin-2-il-vinil)-1H-indazol-6-ilsulfanil]-benzamida. La fórmula molecular es C22H18N4OS y el peso molecular es de 386,47 daltons. La estructura química está representada por la siguiente figura:
Axitinib es un polvo de color blanco a amarillo pálido, con un pKa de 4,8. La solubilidad de axitinib en medios acuosos con un pH que oscila entre 1,1 y 7,8 está por encima de los 0,2 mg/ml. El coeficiente de partición (n-octanol/agua) es de 3,5. INLYTA se suministra como comprimidos recubiertos de color rojo que contienen 1 mg o 5 mg de axitinib junto con celulosa microcristalina, lactosa monohidrato, croscarmelosa sódica, estearato de magnesio y Opadry II rojo 32K15441 como principios inactivos. La cubierta Opadry II 32K15441 roja contiene lactosa monohidrato, HPMC 2910/Hipromelosa 15cP, dióxido de titanio, triacetina (glicerol triacetato) y óxido de hierro rojo. Propiedades farmacodinámicas: Mecanismo de acción: Se ha demostrado que axitinib inhibe los receptores de las tirosinas quinasas, incluidos los receptores del factor de crecimiento del endotelio vascular VEGFR-1, VEGFR-2 y VEGFR-3 a concentraciones plasmáticas terapéuticas. Estos receptores están implicados en la angiogénesis patológica, el crecimiento tumoral y la progresión del cáncer. Axitinib in vitro y en modelos de ratón inhibió la supervivencia y proliferación celular del endotelio inducidas por el VEGF. Se demostró que axitinib inhibe el crecimiento tumoral y la fosforilación del VEGFR-2 en modelos de xenoinjerto de tumor en ratones. Farmacodinamia: El efecto de una dosis oral única de INLYTA (5 mg) sobre el intervalo QTc en ausencia y presencia de 400 mg de ketoconazol se evaluó en un estudio cruzado de dos vías, aleatorizado y simple ciego, del que participaron 35 sujetos sanos. No se detectaron cambios importantes en el intervalo QTc medio (es decir, > 20 ms) respecto del placebo hasta 3 horas después de la dosis. No obstante, no pueden descartarse incrementos menores en el intervalo QTc medio (es decir, < 10 ms). Propiedades farmacocinéticas: En el análisis farmacocinético de la población, se recopilaron datos de 17 ensayos realizados en sujetos sanos y en pacientes con cáncer. Un modelo de distribución bicompartimental con absorción de primer orden y latencias describe de forma adecuada el perfil del axitinib en cuanto a concentración-tiempo. Absorción: Después de la administración de una dosis oral única de 5 mg, la mediana de Tmáx varió entre 2,5 y 4,1 horas. Basado en la vida media plasmática, el estado de equilibrio se espera dentro de los 2 a 3 días de administración de la dosis. La administración de 5 mg de axitinib dos veces al día produjo una acumulación aproximada de 1,4 veces en comparación con la administración de una dosis única. En estado de equilibrio, axitinib exhibe farmacocinética aproximadamente lineal dentro de un rango de dosis de 1 mg a 20 mg. La biodisponibilidad absoluta media de axitinib después de una dosis oral de 5 mg es de 58%. Comparado con un ayuno de toda la noche, la administración de INLYTA junto con una comida de contenido graso moderado produjo un descenso de 10% en el AUC mientras que, con una comida de alto contenido graso y calórico, el aumento del AUC fue de 19%. INLYTA puede administrarse junto con las comidas o lejos de ellas [ver Dosificación]. Distribución: Axitinib muestra una fuerte unión ( > 99%) a las proteínas plasmáticas humanas, con preferencia a la albúmina, y una unión moderada a la alfa-1 glucoproteína ácida. En pacientes con CCR avanzado (n=20), para una dosis de 5 mg dos veces al día con las comidas, la media geométrica (CV%) para Cmáx y AUC0-24 fue de 27,8 (79%) ng/ml y 265 (77%) ng.h/ml, respectivamente. La media geométrica (CV%) para la depuración y el volumen aparente de distribución fue de 38 (80%) l/h y 160 (105%) l, respectivamente. Metabolismo: La vida media plasmática de INLYTA varía entre 2,5 y 6,1 horas. Axitinib se metaboliza principalmente en el hígado a través del CYP3A4/5 y, en menor medida, de CYP1A2, CYP2C19 y UGT1A1. Eliminación: Después de la administración oral de una dosis radiactiva de 5 mg de axitinib, el 41% de la radiactividad se recuperó en las heces y el 23% en la orina. El principal componente identificado en las heces fue el axitinib no modificado, que representó un 12% de la dosis. En orina, no se detectó axitinib no modificado y la mayor parte de la radiactividad recuperada consistió en ácido carboxílico y metabolitos sulfóxidos. En plasma, el componente radiactivo predominante (50% de la radiactividad circulante) fue el metabolito N - glucurónido, mientras que el axitinib no modificado y el metabolito sulfóxido representaron cada uno alrededor del 20% de la radiactividad circulante. Los metabolitos sulfóxidos y N-glucurónidos muestran una potencia in vitro aproximadamente ≥400 veces más baja contra el VEGFR-2 en comparación con axitinib. Interacciones medicamentosas: Efectos de otros medicamentos sobre INLYTA: Axitinib se metaboliza principalmente en el hígado mediante el CYP3A4/5. Además, la solubilidad acuosa de axitinib depende del pH: a mayor pH, menor solubilidad. La figura 1 describe los efectos de un potente inhibidor del CYP3A4/5, un potente inductor del CYP3A4/5 y un antiácido sobre la farmacocinética de axitinib [ver Dosificación e Interacciones].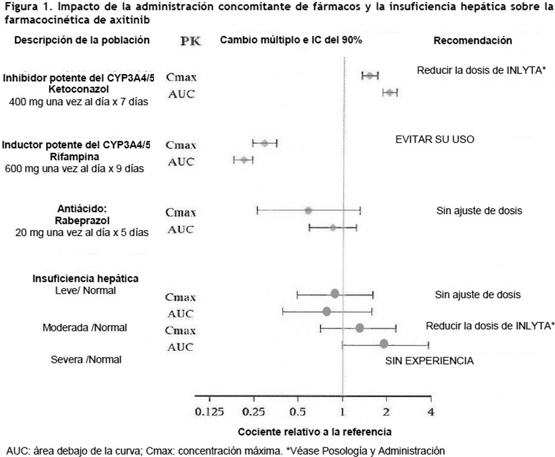
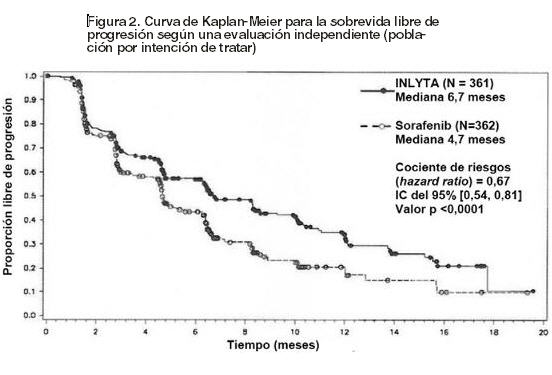
Efectos de INLYTA sobre otros medicamentos: Los estudios in vitro demostraron que axitinib tiene el potencial de inhibir el CYP1A2 y el CYP2C8. Sin embargo, la coadministración de axitinib con paclitaxel, un sustrato del CYP2C8, no incrementó las concentraciones plasmáticas del paclitaxel en los pacientes. Los estudios in vitro indicaron que, a concentraciones plasmáticas terapéuticas, axitinib no inhibe el CYP2A6, CYP2C9, CYP2C19, CYP2D6, CYP2E1, CYP3A4/5 o UGT1A1. Los estudios in vitro en hepatocitos humanos indicaron que axitinib no induce al CYP1A1, CYP1A2 o CYP3A4/5. Axitinib es un inhibidor del transportador de eflujo P-glucoproteína (P-gp) in vitro. Sin embargo, esta inhibición no es esperable a concentraciones plasmáticas terapéuticas. Farmacocinética en poblaciones especiales: Uso pediátrico: no se llevaron a cabo estudios de INLYTA en pacientes menores de 18 años. Deterioro de la función hepática: los efectos del deterioro de la función hepática sobre la farmacocinética de axitinib se describen en la figura 1 [ver Dosificación, Advertencias y Precauciones y Uso en poblaciones específicas]. Deterioro de la función renal: el análisis farmacocinético poblacional (basado en la función renal preexistente) se llevó a cabo en 590 voluntarios sanos y pacientes, incluidos cinco pacientes con deterioro de la función renal severo (15 ml/min ≤ CLcr < 29 ml/min), 64 pacientes con deterioro de la función renal moderado (30 ml/min ≤ CLcr < 59 ml/min), y 139 pacientes con deterioro de la función renal leve (60 ml/min ≤ CLcr < 89 ml/min). El deterioro de la función renal leve a severo no tuvo efectos significativos sobre la farmacocinética de axitinib. Sólo se dispone de los datos de un paciente con enfermedad renal en etapa terminal [ver Uso en poblaciones específicas]. Otros factores intrínsecos: los análisis farmacocinéticos poblacionales indicaron que la edad, el sexo, la raza, el peso corporal, la superficie corporal, el genotipo UGT1A1 y el genotipo CYP2C19 no producen efectos clínicamente relevantes sobre la depuración de axitinib. Toxicología no clínica: Carcinogénesis, mutagénesis, trastornos de fertilidad: No se han realizado estudios de carcinogenicidad con axitinib. Axitinib no mostró respuesta mutagénica en un ensayo (Ames) de mutación inversa bacteriana in vitro ni respuesta clastogénica en un ensayo de aberración cromosómica in vitro con linfocitos humanos. Axitinib tuvo efectos genotóxicos en un ensayo de micronúcleos in vivo con médula ósea de ratón. INLYTA tiene el potencial de deteriorar la función reproductiva y la fertilidad en humanos. En estudios toxicológicos de dosis repetidas, el deterioro del aparato reproductor masculino se observó en los testículos y el epidídimo (disminución del peso del órgano, atrofia o degeneración, disminución de la cantidad de células germinativas, hipospermia o espermatozoides con formas anormales, disminución en la densidad y el recuento de espermatozoides) a dosis ≥15 mg/kg administradas dos veces al día en ratones (aproximadamente 7 veces la exposición sistémica (AUC) de los pacientes a la dosis inicial recomendada) y dosis ≥1,5 mg/kg administradas dos veces al día en perros (aproximadamente 0,1 veces el AUC de los pacientes a la dosis inicial recomendada). Los hallazgos del sistema reproductor femenino en ratones y perros incluyeron signos de retraso de la maduración sexual, reducción o ausencia del cuerpo lúteo, disminución de los pesos uterinos y atrofia uterina a ≥5 mg/kg/dosis (aproximadamente 1,5 o 0,3 veces el AUC de los pacientes a la dosis inicial recomendada en comparación con ratones y perros, respectivamente). En un estudio de fertilidad con ratones, axitinib no afectó ni el apareamiento ni la tasa de fertilidad en los machos cuando se lo administró por vía oral dos veces al día en cualquiera de las dosis evaluadas hasta 50 mg/kg/dosis después de al menos 70 días de administración (aproximadamente 57 veces el AUC de los pacientes a la dosis inicial recomendada). En ratones hembra, se observó disminución de la fertilidad y la viabilidad embrionaria a todas las dosis evaluadas (≥15 mg/kg/dosis administrados por vía oral dos veces al día) después de al menos 15 días de tratamiento con axitinib (aproximadamente 10 veces el AUC de los pacientes a la dosis inicial recomendada). Ensayos clínicos: En un estudio de fase 3, aleatorizado, abierto y multicéntrico, se evaluó la seguridad y eficacia de INLYTA. Los pacientes (N=723) con CCR avanzado cuya enfermedad había progresado durante o después de 1 tratamiento previo con terapias sistémicas (como regímenes que contenían sunitinib, bevacizumab, temsirolimus o citoquinas) fueron aleatorizados (1:1) para recibir INLYTA (N=361) o sorafenib (N=362). La sobrevida libre de progresión (SLP) fue evaluada por un comité de revisión central, independiente y en ciego. Otras variables estudiadas fueron las tasas de respuesta objetiva (ORR, por sus siglas en inglés) y la sobrevida global (SG). Del total de inscriptos en este estudio, 389 pacientes (54%) habían recibido un tratamiento previo basado en sunitinib; 251 pacientes (35%), un tratamiento previo basado en citoquinas (interleucina-2 o interferón- alfa); 59 pacientes (8%), un tratamiento previo basado en bevacizumab y 24 pacientes (3%), un tratamiento previo basado en temsirolimus. Las características demográficas y patológicas iniciales fueron similares entre los grupos tratados con INLYTA y sorafenib respecto de la edad (mediana de 61 años), el sexo (72% masculinos), la raza (75% blancos, 21% asiáticos), el estado funcional del Grupo Oncológico Cooperativo del Este (ECOG performance status, por sus siglas en inglés) (ECOG 0, 55%; ECOG 1, 45%) y la histología (99% de células claras). Se observó una ventaja estadísticamente significativa de INLYTA respecto de sorafenib para la SLP (véase la tabla 3 y la figura 2), sin ninguna diferencia estadísticamente significativa para la SG.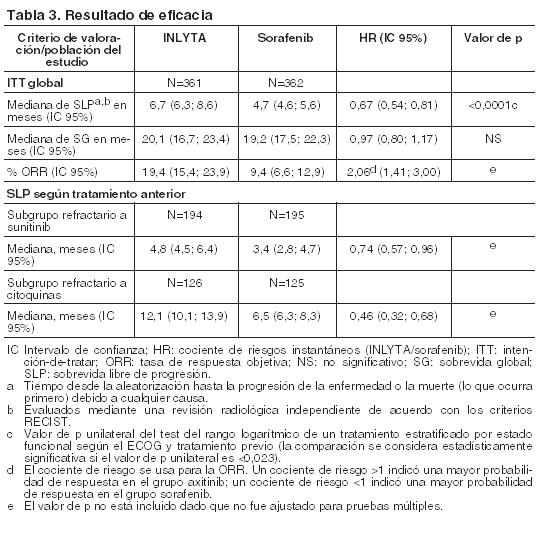
Indicaciones.
INLYTA está indicado para el tratamiento del carcinoma de células renales (CCR) avanzado luego del fracaso de un tratamiento sistémico previo.
Dosificación.
Posología recomendada: La dosis oral inicial recomendada de INLYTA es de 5 mg dos veces al día. Administrar las dosis de INLYTA con una separación de aproximadamente 12 horas con o sin alimentos [ver Farmacología clínica]. INLYTA debe ingerirse entero con un vaso de agua. No se debe administrar una dosis adicional si el paciente vomita u omite una dosis. La siguiente dosis indicada se debe tomar en el horario habitual. Lineamientos para la modificación de la dosis: El aumento o la reducción de la dosis se recomienda en función de la seguridad y la tolerabilidad individuales. Durante el curso del tratamiento, pueden aumentar la dosis los pacientes que toleran INLYTA durante por lo menos dos semanas consecutivas sin reacciones adversas > grado 2 [según los Criterios Comunes de Toxicidad para Eventos Adversos (CTCAE, por sus siglas en inglés)] son normotensos y no reciben medicación antihipertensiva. Cuando se recomienda un aumento de la dosis de 5 mg dos veces al día, la dosis de INLYTA puede aumentarse a 7 mg dos veces al día y posteriormente a 10 mg dos veces al día usando el mismo criterio. Durante el curso del tratamiento, el abordaje de algunas reacciones adversas puede requerir una interrupción temporaria o una discontinuación permanente o una reducción de la dosis de INLYTA [ver Advertencias]. Si se requiere una reducción de la dosis de 5 mg dos veces al día, la dosis recomendada es de 3 mg dos veces al día. Si se requiere otra reducción de la dosis, la dosis recomendada es de 2 mg dos veces al día. Inhibidores potentes del CYP3A4/5: debe evitarse el uso concomitante de inhibidores potentes del CYP3A4/5 (por ej. ketoconazol, itraconazol, claritromicina, atazanavir, indinavir, nefazodona, nelfinavir, ritonavir, saquinavir, telitromicina y voriconazol). Se recomienda la selección de un medicamento concomitante alternativo que no tenga o que tenga un potencial mínimo de inhibición del CYP3A4/5. Aunque el ajuste de dosis de INLYTA no se ha estudiado en pacientes que reciben inhibidores potentes del CYP3A4/5, si se debe administrar concomitantemente un inhibidor potente del CYP3A4/5, se recomienda una disminución de la dosis de INLYTA en aproximadamente la mitad, ya que se prevé que esta reducción ajustará el área bajo la curva de la concentración plasmática de axitinib versus tiempo (AUC) al rango observado sin inhibidores. Las dosis posteriores pueden aumentarse o disminuirse según la seguridad y tolerabilidad individual. Si se suspende la administración concomitante del inhibidor potente, la dosis de INLYTA debe retornar (luego de 3-5 vidas medias del inhibidor) a la utilizada antes de comenzar con el inhibidor potente del CYP3A4/5 [ver Interacciones y Farmacología]. Deterioro de la función hepática: no se requiere ningún ajuste de la dosis inicial cuando se administra INLYTA a pacientes con deterioro de la función hepática leve (Child-Pugh clase A). Con base en los datos farmacocinéticos, la dosis inicial de INLYTA debe reducirse en aproximadamente la mitad en pacientes con deterioro de la función hepática moderado inicial (Child-Pugh clase B). Las dosis posteriores pueden aumentarse o disminuirse con base en la seguridad y tolerabilidad individual. No se ha estudiado INLYTA en pacientes con deterioro de la función hepática severo (Child-Pugh clase C) [ver Advertencias, Uso en poblaciones específicas y Farmacología].
Contraindicaciones.
El uso de INLYTA está contraindicado en pacientes con hipersensibilidad al axitinib o a algún otro componente de INLYTA.
Reacciones adversas.
Dado que los estudios clínicos se llevan a cabo bajo condiciones ampliamente variables, las tasas de reacciones adversas observadas en los estudios clínicos de un medicamento no pueden compararse directamente con las tasas observadas en los estudios clínicos de otro medicamento y no siempre reflejan las tasas observadas en la práctica clínica. Se evaluó la seguridad de INLYTA en 715 pacientes que participaron en estudios con monoterapia, de los cuales 537 presentaban un CCR avanzado. Los datos descriptos [ver Reacciones adversas] reflejan exposición a INLYTA en 359 pacientes con CCR avanzado que participaron en un estudio clínico aleatorizado de comparación con sorafenib [ver Estudios clínicos]. Los siguientes riesgos, así como las medidas adecuadas que deben tomarse, se analizan con más detalle en otras secciones del prospecto: hipertensión, eventos tromboembólicos arteriales, eventos tromboembólicos venosos, hemorragia, insuficiencia cardíaca, perforación gastrointestinal y formación de fístulas, disfunción tiroidea, complicaciones en la cicatrización de las heridas, SLPR, proteinuria, elevación de las enzimas hepáticas, deterioro de la función hepática y anomalías en el desarrollo fetal [ver Advertencias]. Experiencia de estudios clínicos: La mediana de la duración del tratamiento fue de 6,4 meses (rango: 0,03 a 22,0) para los pacientes que recibieron INLYTA y de 5,0 meses (rango: 0,03 a 20,1) para los pacientes que recibieron sorafenib. Se indicaron modificaciones de dosis o retraso temporal del tratamiento debido a una reacción adversa en 199 de los 359 pacientes (55%) que recibieron INLYTA y 220 de los 355 pacientes (62%) que recibieron sorafenib. Se informó discontinuación permanente debido a reacción adversa en 34 de los 359 pacientes (9%) tratados con INLYTA y 46 de los 355 pacientes (13%) tratados con sorafenib. Las reacciones adversas más frecuentes (≥20%) observadas después del tratamiento con INLYTA fueron diarrea, hipertensión, fatiga, disminución del apetito, náuseas, disfonía, síndrome de eritrodisestesia palmo-plantar (mano-pie), pérdida de peso, vómitos, astenia y constipación. La tabla 1 detalla las reacciones adversas informadas en ≥10% de los pacientes que recibieron INLYTA o sorafenib.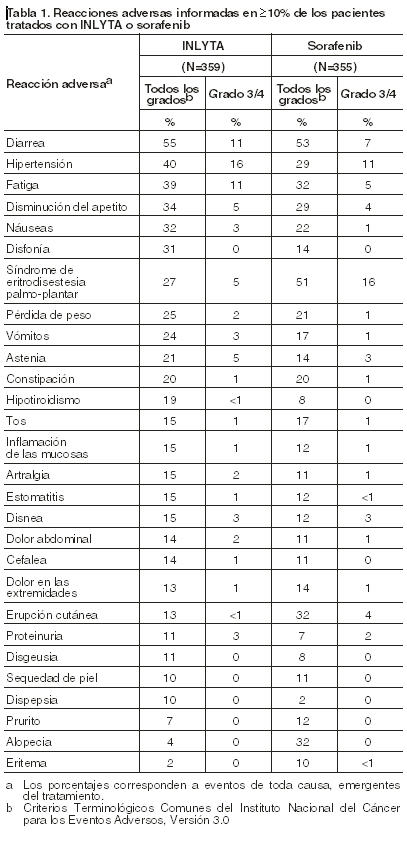
Las reacciones adversas específicas (de todos los grados) que fueron informadas en < 10% de los pacientes tratados con INLYTA incluyeron mareos (9%), dolor abdominal superior (8%), mialgia (7%), deshidratación (6%), epistaxis (6%), anemia (4%), hemorroides (4%), hematuria (3%), acúfenos (3%), elevación de la lipasa (3%), glosodinia (3%), embolia pulmonar (2%), hemorragia rectal (2%), hemoptisis (2%), trombosis venosa profunda (1%), trombosis/oclusión venosa-retiniana (1%), policitemia (1%), y accidente isquémico transitorio (1%). La tabla 2 presenta las alteraciones de laboratorio más frecuentes informadas en ≥10% de los pacientes que recibieron INLYTA o sorafenib.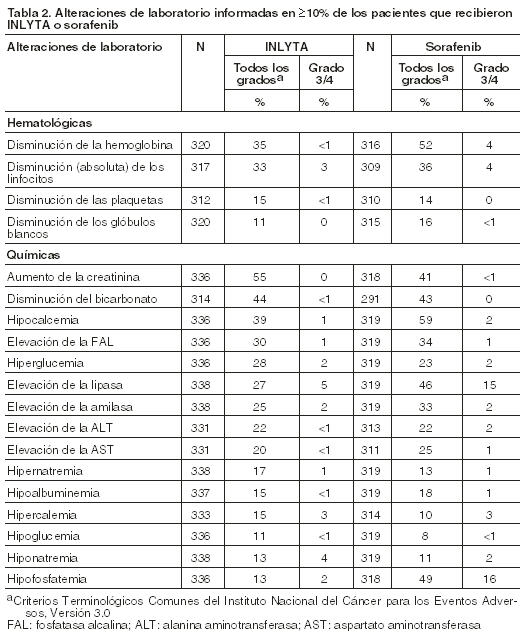
Las alteraciones de laboratorio específicas (de todos los grados) informadas en < 10% de los pacientes tratados con INLYTA incluyeron elevación de la hemoglobina (por encima del límite superior de la normalidad) (9% para INLYTA versus 1% para sorafenib) e hipercalemia (6% para INLYTA frente al 2% para sorafenib).
Precauciones.
Interacción con otros medicamentos y otras formas de interacción: Los datos in vitro indican que axitinib se metaboliza principalmente por vía del CYP3A4/5 y, en menor medida, del CYP1A2, el CYP2C19 y la uridina difosfato-glucuronosiltransferasa 1A1 (UGT1A1). Inhibidores del CYP3A4/5: La coadministración de ketoconazol, un potente inhibidor del CYP3A4/5, incrementó la exposición plasmática de axitinib en voluntarios sanos. Debe evitarse la coadministración de INLYTA con inhibidores potentes del CYP3A4/5. El pomelo o el jugo de pomelo también pueden incrementar las concentraciones plasmáticas de axitinib, por lo que deben evitarse. Se recomienda seleccionar una medicación concomitante cuyo potencial de inhibir el CYP3A4/5 sea mínimo o nulo. Si la coadministración de un inhibidor potente del CYP3A4/5 es imprescindible, debe reducirse la dosis de INLYTA [ver Dosificación y Farmacología clínica]. Inductores del CYP3A4/5: La coadministración de rifampina, un potente inductor del CYP3A4/5, redujo la exposición plasmática de axitinib en voluntarios sanos. Debe evitarse la coadministración de INLYTA con potentes inductores del CYP3A4/5 (por ej., rifampina, dexametasona, fenitoína, carbamazepina, rifabutina, rifapentina, fenobarbital e hipérico). Se recomienda seleccionar una medicación concomitante cuyo potencial de inducir el CYP3A4/5 sea mínimo o nulo [ver Dosificación y Farmacología]. Los inductores moderados del CYP3A4/5 (por ej., bosentán, efavirenz, etravirina, modafinilo y nafcilina) también pueden reducir la exposición plasmática de axitinib, por lo que debe evitarse, de ser posible. Embarazo y lactancia: Embarazo categoría D: No se han llevado a cabo estudios adecuados y bien controlados de INLYTA en mujeres embarazadas. El mecanismo de acción de INLYTA puede causar daño fetal cuando se lo administra durante el embarazo. En ratones, axitinib mostró efectos teratogénicos, embriotóxicos y fetotóxicos a exposiciones inferiores a las exposiciones humanas a las dosis de inicio recomendadas. Si este medicamento se administra durante el embarazo o si el embarazo se produce durante el tratamiento con este medicamento, la paciente debe estar informada acerca de los riesgos potenciales para el feto. Axitinib oral administrado dos veces al día a ratones hembra antes del apareamiento y durante la primera semana de embarazo causó un aumento en la pérdida post-implantación a todos los niveles de dosis evaluados (≥15 mg/kg/dosis, aproximadamente 10 veces la exposición sistémica (AUC) de los pacientes a la dosis inicial recomendada). En un estudio de toxicidad en el desarrollo embrionario-fetal, las hembras preñadas de ratón recibieron dosis orales de 0,15, 0,5 y 1,5 mg/kg/dosis de axitinib dos veces al día durante el período de organogénesis. Las toxicidades embrio-fetales observadas en ausencia de toxicidad materna incluyeron malformaciones (paladar hendido) a dosis de 1,5 mg/kg (aproximadamente 0,5 veces el AUC de los pacientes a la dosis inicial recomendada) y variaciones en la osificación de los huesos a una dosis ≥0,5 mg/kg (aproximadamente 0,15 veces el AUC de las pacientes a la dosis inicial recomendada). Lactancia: Se desconoce si el axitinib se excreta en la leche materna. Dado que muchos medicamentos se excretan en la leche materna y debido al potencial de reacciones adversas serias de INLYTA en lactantes, es necesario decidir si se interrumpe la lactancia o la administración del medicamento, teniendo en cuenta la importancia de este para la madre. Uso pediátrico: No se han llevado a cabo estudios sobre la seguridad y la eficacia de INLYTA en pacientes pediátricos. Se observaron toxicidades en los huesos y los dientes de ratones y perros inmaduros que recibieron axitinib oral dos veces al día durante 1 mes o más. En ratones y perros, los efectos óseos consistieron en engrosamiento de la placa de crecimiento a dosis ≥15 mg/kg (aproximadamente 6 y 15 veces, respectivamente, la exposición sistémica (AUC) de los pacientes a la dosis inicial recomendada). Se observaron anomalías de crecimiento en los incisivos (como caries dentales, maloclusiones y roturas o faltas dentarias) de ratones a los que se administró axitinib oral dos veces al día en dosis ≥5 mg/kg (aproximadamente 1,5 veces el AUC de los pacientes a la dosis inicial recomendada). No se estudiaron otras toxicidades de importancia potencial para los pacientes pediátricos en animales jóvenes. Uso geriátrico: En un estudio clínico controlado con INLYTA para el tratamiento de pacientes con CCR, 123 de los 359 pacientes (34%) tratados con INLYTA tenían 65 años o más. Aunque no se puede descartar una mayor sensibilidad en algunos individuos mayores, la seguridad y eficacia de INLYTA no mostró diferencias generales entre los pacientes ≥65 años y los pacientes más jóvenes. No se requieren ajustes de dosis en los pacientes ancianos [ver Dosificación y Farmacología]. Deterioro de la función hepática: En un estudio especializado de deterioro de la función hepática comparada a sujetos con función hepática normal, la exposición sistémica posterior a una dosis única de INLYTA resultó similar en individuos con deterioro de la función hepática basal leve (clase A de la escala Child-Pugh) y más elevada en individuos con deterioro de la función hepática basal moderado (clase B de la escala Child-Pugh). No se requiere ajuste de la dosis inicial cuando se administra INLYTA a pacientes con deterioro de la función hepática leve (clase A de la escala Child-Pugh). Se recomienda disminuir la dosis inicial cuando se administra INLYTA a pacientes con deterioro de la función hepática moderado (clase B de la escala Child-Pugh) [ver Dosificación, Advertencias y Farmacología]. No se realizaron estudios con INLYTA en pacientes con deterioro de la función hepática severo (clase C de la escala Child-Pugh). Deterioro de la función renal: No se realizaron estudios especializados sobre deterioro de la función renal con axitinib. Basado en los análisis farmacocinéticos poblacionales, la depuración de axitinib no mostró diferencias significativas en pacientes con deterioro de la función renal leve a severo preexistente (15 ml/min ≤ depuración de creatinina [Clcr] < 89 ml/min) [ver Farmacología clínica]. No se necesita ajuste de la dosis inicial en pacientes con deterioro de la función renal leve a severo preexistente. Debe administrarse con cuidado en pacientes con enfermedad renal en etapa terminal (Clcr < 15 ml/min).
Advertencias.
Hipertensión y crisis hipertensiva: En un estudio clínico controlado con INLYTA para el tratamiento de pacientes con carcinoma de células renales (CCR), se informó hipertensión en 145 de 359 pacientes (40%) tratados con INLYTA y 103 de 355 pacientes (29%) tratados con sorafenib. La hipertensión de grado 3/4 se observó en 56 de 359 pacientes (16%) tratados con INLYTA y 39 de 355 pacientes (11%) tratados con sorafenib, mientras que se informaron crisis hipertensivas en 2 de los 359 pacientes ( < 1%) tratados con INLYTA y en ninguno de los pacientes tratados con sorafenib. El tiempo medio de aparición de la hipertensión (tensión arterial sistólica > 150 mmHg o tensión arterial diastólica > 100 mmHg) se observó dentro del primer mes del inicio del tratamiento con INLYTA y fueron informados aumentos de la tensión arterial apenas 4 días después de iniciado el tratamiento. La hipertensión fue tratada con un tratamiento antihipertensivo estándar. La interrupción del tratamiento con INLYTA debido a hipertensión se produjo en 1 de los 359 pacientes ( < 1%) que recibieron INLYTA y ninguno de los pacientes que recibieron sorafenib [ver Reacciones adversas]. Los niveles de tensión arterial deben estar controlados adecuadamente antes de iniciar el tratamiento con INLYTA. Los pacientes deben ser monitoreados en búsqueda de hipertensión y recibir tratamiento antihipertensivo estándar según se requiera. Si la hipertensión persiste, a pesar del tratamiento antihipertensivo, reducir la dosis de INLYTA. Si, pese al tratamiento antihipertensivo y la reducción de la dosis, la hipertensión continúa siendo severa y persistente, discontinuar la administración de INLYTA. La discontinuación del tratamiento con INLYTA debe considerarse también en caso de crisis hipertensiva. Si se interrumpe el tratamiento con INLYTA, debe controlarse la aparición de hipotensión en los pacientes que reciben medicación antihipertensiva [ver Dosificación]. Eventos tromboembólicos arteriales: En los estudios clínicos, se han informado eventos tromboembólicos arteriales, en algunos casos seguidos de muerte. En un estudio clínico controlado con INLYTA para el tratamiento de pacientes con CCR, se informaron eventos tromboembólicos arteriales de grado 3/4 en 4 de 359 pacientes (1%) tratados con INLYTA y 4 de 355 pacientes (1%) tratados con sorafenib. Se informó un caso de accidente cerebrovascular fatal en 1 de 359 pacientes ( < 1%) tratados con INLYTA y ningún caso entre los pacientes tratados con sorafenib [ver Reacciones adversas]. En los estudios clínicos con INLYTA, los eventos tromboembólicos arteriales (incluidos accidente isquémico transitorio, accidente cerebrovascular, infarto de miocardio y oclusión arterial retiniana) fueron informados en 17 de 715 pacientes (2%), con dos casos de muerte secundaria a accidente cerebrovascular. INLYTA debe usarse con precaución en pacientes con riesgo o antecedentes de cualquiera de estos eventos. INLYTA no se estudió en pacientes que presentaron un evento tromboembólico arterial dentro de los 12 meses anteriores al estudio. Eventos tromboembólicos venosos: En los estudios clínicos, se han informado eventos tromboembólicos venosos, en algunos casos seguidos de muerte. En un estudio clínico controlado con INLYTA para el tratamiento de pacientes con CCR, se informaron eventos tromboembólicos venosos en 11 de 359 pacientes (3%) tratados con INLYTA y 2 de 355 pacientes (1%) tratados con sorafenib. Se informaron eventos tromboembólicos venosos de grado 3/4 en 9 de 359 pacientes (3%) tratados con INLYTA (incluidos embolia pulmonar, trombosis venosa profunda, oclusión venosa retiniana y trombosis venosa retiniana) y 2 de 355 pacientes (1%) tratados con sorafenib. Se informó un caso de embolia pulmonar fatal entre los 359 pacientes ( < 1%) tratados con INLYTA y ningún caso entre los pacientes tratados con sorafenib. En los estudios clínicos con INLYTA, se informaron eventos tromboembólicos venosos en 22 de 715 pacientes (3%) y dos casos de muerte secundaria a embolia pulmonar. INLYTA debe usarse con precaución en pacientes con riesgo o antecedentes de cualquiera de estos eventos. INLYTA no se estudió en pacientes que presentaron un evento tromboembólico venoso dentro de los 6 meses anteriores al estudio. Hemorragia: En un estudio clínico controlado con INLYTA para el tratamiento de pacientes con CCR, se informaron eventos hemorrágicos en 58 de 359 pacientes (16%) tratados con INLYTA y 64 de 355 pacientes (18%) tratados con sorafenib. Se observaron eventos hemorrágicos de grado 3/4 en 5 de 359 pacientes (1%) tratados con INLYTA (incluidos hemorragia cerebral, hematuria, hemoptisis, hemorragia digestiva baja y melena) y 11 de 355 pacientes (3%) tratados con sorafenib. Se informó 1 caso de hemorragia con desenlace fatal entre los 359 pacientes ( < 1%) que recibieron INLYTA (hemorragia gástrica) y 3 casos entre los 355 pacientes (1%) que recibieron sorafenib. INLYTA no se estudió en pacientes con evidencia de metástasis cerebral no tratada o de hemorragia digestiva activa reciente y, por lo tanto, no debería administrarse a dichos pacientes. Si la hemorragia requiere intervención médica, la administración de INLYTA debe interrumpirse temporalmente. Insuficiencia cardíaca: En un estudio clínico controlado con INLYTA para el tratamiento de pacientes con CCR, se informó insuficiencia cardíaca en 6 de 359 pacientes (2%) tratados con INLYTA y 3 de 355 pacientes (1%) tratados con sorafenib. Se observó insuficiencia cardíaca de grado 3/4 en 2 de 359 pacientes (1%) tratados con INLYTA y 1 de 355 pacientes ( < 1%) tratados con sorafenib. Se informó insuficiencia cardíaca fatal en 2 de 359 pacientes (1%) tratados con INLYTA y 1 de 355 pacientes ( < 1%) tratados con sorafenib. Los signos o síntomas de insuficiencia cardíaca deben controlarse a lo largo del tratamiento con INLYTA. El manejo de la insuficiencia cardíaca puede requerir la discontinuación de forma permanente del tratamiento con INLYTA. Perforación gastrointestinal y formación de fístulas: En un estudio clínico controlado con INLYTA para el tratamiento de pacientes con CCR, se informó perforación gastrointestinal en 1 de 359 pacientes ( < 1%) tratados con INLYTA y ninguno de los pacientes tratados con sorafenib. En los estudios clínicos con INLYTA, se informó perforación gastrointestinal en 5 de 715 pacientes (1%), con un caso de desenlace fatal. Además de los casos de perforación gastrointestinal, se informó formación de fístulas en 4 de los 715 pacientes (1%). La aparición de síntomas de perforación gastrointestinal o fístulas debe controlarse periódicamente a lo largo del tratamiento con INLYTA. Disfunción tiroidea: En un estudio clínico controlado con INLYTA para el tratamiento de pacientes con CCR, se informó hipotiroidismo en 69 de 359 pacientes (19%) tratados con INLYTA y 29 de 355 pacientes (8%) tratados con sorafenib e hipertiroidismo en 4 de 359 pacientes (1%) tratados con INLYTA y 4 de 355 pacientes (1%) tratados con sorafenib. En pacientes que presentaron niveles de TSH (hormona estimulante de la tiroides) < 5 mU/ml antes del tratamiento, se informaron elevaciones de la TSH a ≥10 mU/ml en 79 de 245 pacientes (32%) que recibieron INLYTA y 25 de 232 pacientes (11%) que recibieron sorafenib [ver Reacciones adversas]. La función tiroidea debe controlarse antes de la iniciación y durante el transcurso del tratamiento con INLYTA. El hipotiroidismo y el hipertiroidismo deben tratarse según la práctica médica estándar de conservación del estado eutiroideo. Complicaciones en la cicatrización de heridas: No se llevaron a cabo estudios formales sobre el efecto de INLYTA en la cicatrización de las heridas. El tratamiento con INLYTA debe interrumpirse al menos 24 horas antes de una intervención quirúrgica programada. La decisión de retomar el tratamiento con INLYTA después de la cirugía debe basarse en los criterios clínicos de una cicatrización adecuada de la herida. Síndrome de leucoencefalopatía posterior reversible: En un estudio clínico controlado con INLYTA para el tratamiento de pacientes con CCR, se informó síndrome de leucoencefalopatía posterior reversible (SLPR) en 1 de 359 pacientes ( < 1%) tratados con INLYTA y ninguno de los pacientes tratados con sorafenib [ver Reacciones adversas]. Se informaron dos casos adicionales de SLPR en otros estudios clínicos con INLYTA. El SLPR es un trastorno neurológico que puede presentarse con cefalea, convulsiones, letargo, confusión, ceguera y otros trastornos visuales o neurológicos. También puede presentarse hipertensión leve a severa. El diagnóstico de SLPR se confirma mediante resonancia magnética. Debe discontinuarse la administración de INLYTA en pacientes que desarrollan SLPR. Se desconoce la seguridad de reiniciar el tratamiento con INLYTA en pacientes que han experimentado SLPR con anterioridad. Proteinuria: En un estudio clínico controlado con INLYTA para el tratamiento de pacientes con CCR, se informó proteinuria en 39 de los 359 pacientes (11%) tratados con INLYTA y 26 de los 355 pacientes (7%) tratados con sorafenib. Se informó proteinuria de grado 3 en 11 de los 359 pacientes (3%) que recibieron INLYTA y 6 de los 355 pacientes (2%) que recibieron sorafenib [ver Reacciones adversas]. Se recomienda monitorear la presencia de proteinuria antes de la iniciación y durante el transcurso del tratamiento con INLYTA en forma periódica. En pacientes que desarrollan proteinuria moderada a severa, reducir la dosis o interrumpir temporalmente el tratamiento con INLYTA. Elevación de las enzimas hepáticas: En un estudio clínico controlado con INLYTA para el tratamiento de pacientes con CCR, se informaron elevaciones de la alanina aminotransferasa (ALT) de todos los grados en el 22% de los pacientes para ambos grupos de tratamiento, con eventos de grado 3/4 en < 1% de los pacientes tratados con INLYTA y 2% de los pacientes tratados con sorafenib. Los niveles de alanina aminotransferasa (ALT), de aspartato aminotransferasa (AST) y de bilirrubina deben monitorearse antes de comenzar el tratamiento con INLYTA y periódicamente durante su transcurso. Deterioro de la función hepática: La exposición sistémica a axitinib fue superior en individuos con deterioro moderado de la función hepática (clase B de la escala Child-Pugh) en comparación con los individuos cuya función hepática era normal. Se recomienda disminuir la dosis de INLYTA en pacientes con deterioro moderado de la función hepática (clase B de la escala Child-Pugh). INLYTA no se estudió en pacientes con deterioro de la función hepática severo (clase C de la escala Child-Pugh) [ver Dosificación, Uso en poblaciones específicas y Farmacología]. Embarazo: En pacientes embarazadas, el mecanismo de acción de INLYTA puede causar daño fetal. No se cuenta con estudios clínicos adecuados y bien controlados en pacientes embarazadas en tratamiento con INLYTA. En estudios de toxicidad del desarrollo en ratones, axitinib mostró efectos teratogénicos, embriotóxicos y fetotóxicos a exposiciones maternas inferiores a las exposiciones humanas a las dosis clínicas recomendadas. Debe aconsejarse a las mujeres en edad fértil que eviten embarazarse mientras reciben INLYTA. Las mujeres embarazadas tratadas con este medicamento o que se embarazan durante el tratamiento deben recibir información acerca de los riesgos potenciales para el feto [ver Uso en poblaciones específicas].
Conservación.
Conservar a temperatura entre 20-25 °C; con excursiones permitidas entre 15 °C a 30 °C. Almacenar en su envase original.
Sobredosificación.
No existe tratamiento específico para la sobredosis de INLYTA. En un estudio clínico controlado con INLYTA para el tratamiento de pacientes con CCR, se informaron mareos (grado 1) en un paciente que recibió una dosis de 20 mg dos veces al día durante 4 días por error. En un estudio clínico de determinación de dosis con INLYTA, los individuos que recibieron dosis iniciales de 10 mg dos veces al día o de 20 mg dos veces al día experimentaron reacciones adversas que incluyeron hipertensión, convulsiones asociadas a hipertensión y hemoptisis fatal. Ante la sospecha de sobredosis, suspender INLYTA e instituir medidas de soporte. Ante la eventualidad de una sobredosificación, concurrir al Hospital más cercano o comunicarse con los Centros de Toxicología: Hospital de Pediatría Ricardo Gutiérrez: (011) 4962-6666/2247. Hospital A. Posadas: (011) 4658-7777 / 4654-6648.
Información al paciente.
Qué es INLYTA y para qué se utiliza: Inlyta es un medicamento de venta bajo receta, utilizado para el tratamiento de pacientes con cáncer de riñón avanzado [carcinoma de células renales (CCR) avanzado] cuando un tratamiento previo con otro medicamento para dicha enfermedad no funcionó. Se desconoce si INLYTA es seguro o efectivo en niños. Antes de usar INLYTA: Antes de usar INLYTA debería informarle a su médico si: tiene hipertensión arterial, tiene problemas de tiroides, tiene problemas de hígado, tiene antecedentes de coágulos en sus venas o arterias (tipos de vasos sanguíneos), incluyendo accidente cerebrovascular, ataque cardíaco, o cambios en la visión, tiene cualquier problema de sangrado, tiene antecedentes de insuficiencia cardíaca, tiene una herida no cicatrizada, tiene una cirugía programada. Debería dejar de tomar INLYTA por lo menos 24 horas antes de la cirugía planificada, tiene alguna otra condición médica. En el caso de las mujeres, debería informarle a su médico si: está embarazada o planea estarlo. Tomar INLYTA durante su embarazo puede causar la muerte de su bebé o defectos de nacimiento. No debería