JAKAVI®
NOVARTIS
Inhibidor de proteína quinasa.
Composición.
Cada comprimido de Jakavi® 5 mg contiene: Ruxolitinib (correspondiente a 6,60 mg de fosfato de ruxolitinib) 5 mg. Excipientes: lactosa monohidratada 71,45 mg; celulosa microcristalina 68,35mg; carboximetilalmidón de sodio 4,80 mg; hidroxipropilcelulosa 3,20 mg; povidona 3,20 mg; sílice coloidal anhidra 1,60 mg; estearato de magnesio 0,80 mg. Cada comprimido de Jakavi® 15 mg contiene: Ruxolitinib (correspondiente a 19,80 mg de fosfato de ruxolitinib) 15 mg. Excipientes: lactosa monohidratada 214,35 mg; celulosa microcristalina 205,05 mg; carboximetilalmidón de sodio 14,40 mg; hidroxipropilcelulosa 9,60 mg; povidona 9,60 mg; sílice coloidal anhidra 4,80 mg; estearato de magnesio 2,40 mg. Cada comprimido de Jakavi® 20 mg contiene: Ruxolitinib (correspondiente a 26,40 mg de fosfato de ruxolitinib) 20 mg. Excipientes: lactosa monohidratada 285,80 mg; celulosa microcristalina 273,40 mg; carboximetilalmidón de sodio 19,20 mg; hidroxipropilcelulosa 12,80 mg; povidona 12,80 mg; sílice coloidal anhidra 6,40 mg; estearato de magnesio 3,20 mg.
Farmacología.
Mecanismo de acción (MA): Ruxolitinib es un inhibidor selectivo de las quinasas de la familia Jano (JAK) JAK1 y JAK2 (valores de CI50 de 3,3 nM y 2,8 nM para las enzimas JAK1 y JAK2, respectivamente). Dichas quinasas median la transducción de señales iniciada por varias citocinas y factores de crecimiento que son importantes para la hematopoyesis y la función inmunitaria. La transducción de señales vía JAK implica la incorporación de STAT (transductores de señales y activadores de la transcripción) a los receptores de las citocinas y la activación y el traslado posterior de los STAT al núcleo celular, donde modulan la expresión génica. La desregulación de la vía JAK-STAT se ha asociado a diversas neoplasias malignas y a una mayor proliferación y supervivencia de células malignas. Se sabe que la mielofibrosis (MF) es una neoplasia mieloproliferativa (NMP) asociada a la desregulación de la transducción de señales mediada por las enzimas JAK1 y JAK2. Se cree que la base de la desregulación radica en las concentraciones elevadas de citocinas circulantes, que activan la vía JAK-STAT, las mutaciones de ganancia de función, como JAK2V617F, y el silenciamiento de los mecanismos de regulación negativa. Los pacientes con mielofibrosis presentan una desregulación de la transducción de señales mediada por las JAK, con independencia del estado (positivo o negativo) de la mutación JAK2V617F. El ruxolitinib inhibe la vía de transducción de señales JAK-STAT y la proliferación celular en modelos celulares, dependientes de citocinas, de neoplasias malignas hemáticas, así como la proliferación de células Ba/F3 tras volverlas independientes de citocinas mediante la expresión de la proteína mutada JAK2V617F; la CI50 es de entre 80 y 320 nM. En un modelo murino de neoplasia mieloproliferativa portadora de la mutación JAK2V617F, la administración oral de ruxolitinib previno la esplenomegalia, disminuyó preferentemente el número de células portadoras de la mutación JAK2V617F en el bazo, redujo el número de citocinas inflamatorias circulantes (p. ej., TNF-a, IL-6) y dio lugar a una supervivencia murina significativamente mayor en dosis que no produjeron efectos mielodepresores. Propiedades farmacodinámicas: Ruxolitinib inhibe la fosforilación de STAT3, inducida por citocinas, en la sangre de los sujetos sanos y pacientes con mielofibrosis. Dos horas después de administrar el medicamento se logra la inhibición máxima de la fosforilación de STAT3, la cual regresa a los niveles iniciales hacia las 8 horas, tanto en los sujetos sanos como en los pacientes con mielofibrosis, lo cual indica que no se produce una acumulación de compuesto original ni de metabolitos activos. En los sujetos con mielofibrosis, las elevaciones iniciales de los marcadores de la inflamación asociados a síntomas generales, como el TNF alpha, la IL-6 y la CRP, disminuyeron después del tratamiento con ruxolitinib. Los pacientes con mielofibrosis no se volvieron resistentes a los efectos farmacodinámicos del tratamiento con ruxolitinib con el paso del tiempo. En un estudio minucioso del QT en sujetos sanos, no hubo indicios de un efecto prolongador del QT/QTc cuando se administraron dosis únicas de hasta 200 mg de ruxolitinib (que es una dosis supra-terapéutica), lo cual indica que el ruxolitinib carece de efectos sobre la repolarización cardíaca. Ensayos clínicos: Se realizaron 2 estudios aleatorizados de Fase III (COMFORT-I y COMFORT-II) en pacientes con mielofibrosis (ya sea primaria o bien secundaria a policitemia vera o a trombocitemia idiopática). En ambos estudios, los pacientes tenían esplenomegalias palpables de al menos 5 cm por debajo del reborde costal y una categoría de riesgo intermedio 2 (2 factores pronósticos) o de riesgo elevado (3 o más factores pronósticos), según los Criterios Consensuados del Grupo de Trabajo Internacional (IWG). Los factores pronósticos que abarcaban dichos criterios eran: edad > 65 años, presencia de síntomas generales (disminución del peso, fiebre, sudores nocturnos) anemia (hemoglobina < 10 g/dL), leucocitosis (antecedentes de cifras leucocíticas > 25 X 109/L) y blastos circulantes ≥1%. La dosis inicial de Jakavi® se basó en la cifra de trombocitos. Los pacientes con cifras trombocíticas de entre 100000 y 200000/mm3 recibieron inicialmente 15 mg de ruxolitinib (Jakavi®) 2 veces al día y los que tenían cifras trombocíticas > 200000/mm3 recibieron inicialmente 20 mg de ruxolitinib (Jakavi®) 2 veces al día. Las dosis se individualizaron según la tolerabilidad y la eficacia: se administraron dosis máximas de 20 mg 2 veces al día a los pacientes con cifras trombocíticas de 100000 a ≤125000/mm3, de 10 mg 2 veces al día a los pacientes con cifras trombocíticas de 75000 a ≤100000/mm3, y de 5 mg 2 veces al día a los pacientes con cifras trombocíticas de 50000 a ≤75000/mm3. COMFORT-I fue un estudio de doble enmascaramiento, aleatorizado y comparativo con placebo, realizado en 309 pacientes que eran resistentes al tratamiento disponible o que no podían recibirlo. Los pacientes recibieron Jakavi® o el correspondiente placebo. El criterio principal de eficacia fue la proporción de sujetos que a la semana 24 presentaban una reducción ≥35% del volumen del bazo con respecto al inicio, determinada mediante IRM o TC. Los criterios secundarios fueron, por ejemplo, la duración del mantenimiento de la reducción ≥35% del volumen del bazo con respecto al inicio, la proporción de pacientes que a la semana 24 presentaban una reducción ≥50% en la puntuación total de síntomas con respecto al inicio -a juzgar por el diario modificado (v. 2.0) del Formulario de Evaluación de los Síntomas de Mielofibrosis (MFSAF)-, la variación de la puntuación total de síntomas a la semana 24 con respecto al inicio -determinada mediante el diario modificado (v. 2.0) del MFSAF- y la supervivencia general. COMFORT-II fue un estudio sin enmascaramiento, aleatorizado, efectuado en 219 pacientes. Los pacientes fueron asignados aleatoriamente a los grupos de Jakavi® o del mejor tratamiento disponible (en proporción 2:1). El mejor tratamiento disponible fue elegido por el investigador de acuerdo con la situación individual de cada paciente. En el grupo del mejor tratamiento disponible, el 47% de los pacientes recibieron hidroxiurea y el 16% de los pacientes tomaron glucocorticoides. El criterio principal de eficacia fue la proporción de pacientes que a la semana 48 presentaban una reducción ≥35% del volumen del bazo con respecto al inicio, determinada mediante IRM o TC. Uno de los criterios secundarios del estudio COMFORT-II fue la proporción de pacientes que a la semana 24 presentaban una reducción ≥35% del volumen del bazo con respecto al inicio, determinada mediante IRM o TC. Otro criterio secundario fue la duración del mantenimiento de la reducción ≥35% con respecto al inicio en los pacientes que respondían al tratamiento. En el estudio COMFORT-I, los datos personales y las características de la enfermedad de los pacientes eran comparables entre los grupos terapéuticos. La edad mediana fue de 68 años, el 61% de los pacientes eran mayores de 65 años y el 54% de ellos, de sexo masculino. La mitad de los pacientes (50%) padecían de mielofibrosis primaria, el 31% sufría de mielofibrosis secundaria a policitemia y el 18% padecía de mielofibrosis secundaria a trombocitemia idiopática. Veintiuno (21%) de los pacientes recibieron transfusiones eritrocíticas durante el período de 8 semanas de reclutamiento para participar en el estudio. La cifra mediana de trombocitos fue de 251000/mm3. El 76% de los pacientes eran portadores de la mutación que codificaba la sustitución V617F en la proteína JAK. Los bazos de los pacientes tenían una longitud mediana palpable de 16 cm. Al inicio del estudio, el 37,4% de los pacientes del grupo de Jakavi® sufrían de anemias de grado 1, el 31,6%, de grado 2 y el 4,5%, de grado 3, mientras que en el grupo del placebo el 35,8% padecían de anemias de grado 1, el 35,1%, de grado 2, el 4,6%, de grado 3, y el 0,7%, de grado 4. Se describió trombocitopenia de grado 1 en el 12,9% de los pacientes del grupo de Jakavi® y en el 13,2% de los pacientes del grupo del placebo. En el estudio COMFORT-II, los datos personales y las características de la enfermedad de los pacientes eran comparables entre los grupos terapéuticos. La edad mediana fue de 66 años, el 52% de los pacientes eran mayores de 65 años y el 57% de ellos, de sexo masculino. El cincuenta y tres por ciento (53%) de los sujetos padecían de mielofibrosis primaria, el 31% sufría de mielofibrosis secundaria a policitemia vera y el 16% padecía de mielofibrosis secundaria a trombocitemia idiopática. El 19% de los pacientes fueron considerados "dependientes de transfusiones" al inicio. Los bazos de los pacientes tenían una longitud mediana palpable de 15 cm. Al inicio del estudio, el 34,2% de los pacientes del grupo de Jakavi® sufrían de anemias de grado 1, el 28,8%, de grado 2 y el 7,5%, de grado 3, mientras que en el grupo del mejor tratamiento disponible el 37% padecieron de anemias de grado 1, el 27,4%, de grado 2, el 13,7%, de grado 3, y el 1,4%, de grado 4. Se describió trombocitopenia de grado 1 en el 8,2% de los pacientes del grupo de Jakavi® y en el 9,6% de los pacientes del grupo del mejor tratamiento disponible. Los análisis de eficacia del criterio principal de los estudios COMFORT-1 y COMFORT-II se presentan en la Tabla 1. En ambos estudios, una proporción significativamente mayor de pacientes del grupo de Jakavi® presentó una reducción ≥35% del volumen del bazo con respecto al inicio en comparación con el placebo (COMFORT-I) o con el mejor tratamiento disponible (COMFORT-II).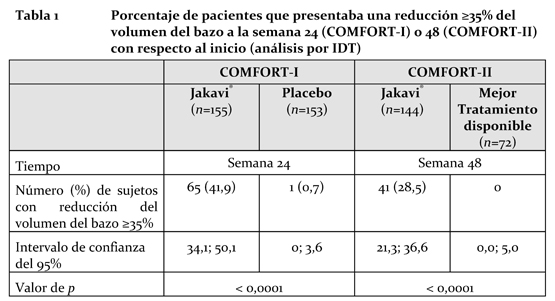
En el estudio COMFORT-I, el 41,9% de los pacientes del grupo de Jakavi® presentaban una reducción ≥35% del volumen del bazo a la semana 24 con respecto al inicio, frente al 0,7% de los pacientes del grupo del placebo. Una proporción similar de pacientes del grupo de Jakavi® presentó una reducción ≥35% en la longitud del bazo palpable. En el estudio COMFORT-II, el 28,5% de los pacientes del grupo de Jakavi® presentó una reducción ≥35% del volumen del bazo a la semana 48 con respecto al inicio, frente a ninguno de los pacientes (0%) del grupo que recibió el mejor tratamiento disponible. Uno de los criterios secundarios fue la proporción de pacientes que presentaban una reducción ≥35% del volumen del bazo a la semana 24 (con respecto al inicio). Una proporción significativamente mayor de pacientes del grupo de Jakavi® igual a 46 (31,9%) presentó dicha reducción, frente a ninguno de los pacientes (0%) del grupo que recibió el mejor tratamiento disponible (valor de p < 0,0001). Una proporción significativamente mayor de pacientes del grupo de Jakavi® presentó una reducción ≥ 35% del volumen del bazo con respecto al inicio, con independencia de la presencia o la ausencia de la mutación JAK2V617F o del sub-tipo de enfermedad (mielofibrosis primaria, mielofibrosis secundaria a policitemia vera o mielofibrosis secundaria a trombocitemia idiopática). La Figura 1 muestra un gráfico en cascada del cambio porcentual, con respecto al inicio, del volumen del bazo a la semana 24 en el estudio COMFORT-I. Entre los 139 pacientes del grupo de Jakavi® en los que se evaluó, al inicio y a la semana 24, el volumen del bazo, todos los pacientes, excepto 2, presentaron algún grado de reducción del volumen del bazo a la semana 24, siendo la reducción mediana del 33%. Entre los 106 pacientes del grupo del placebo en los que se había evaluado el volumen del bazo al inicio y a la semana 24 hubo un aumento mediano del 8,5%.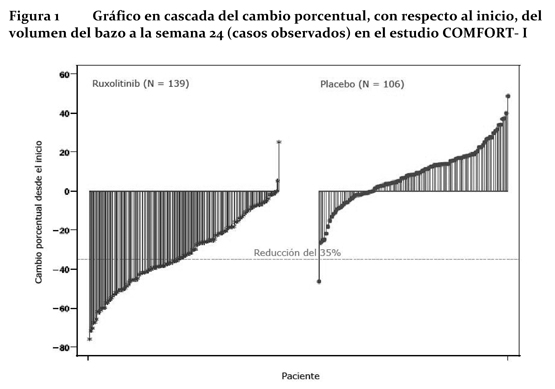
La Figura 2 muestra un gráfico en cascada del cambio porcentual, con respecto al inicio, del volumen del bazo a la semana 48 en el estudio COMFORT-II. Entre los 98 pacientes del grupo de Jakavi® en los que se había evaluado el volumen del bazo al inicio y a la semana 48, la reducción mediana del volumen del bazo a la semana 48 fue del 28%. Entre los 34 pacientes del grupo del mejor tratamiento disponible en los que se había evaluado el volumen del bazo al inicio y a la semana 48, hubo un aumento mediano del 8,5%.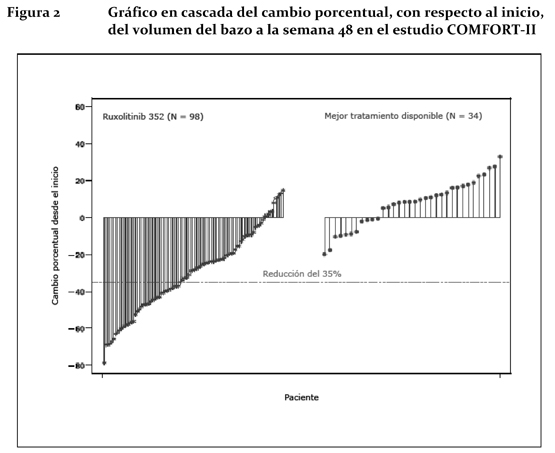
La Tabla 2 muestra la probabilidad de la duración desde la primera reducción ≥35% del volumen del bazo hasta un aumento del 25% con respecto al nadir y la pérdida de la respuesta en los estudios COMFORT-I y COMFORT-II.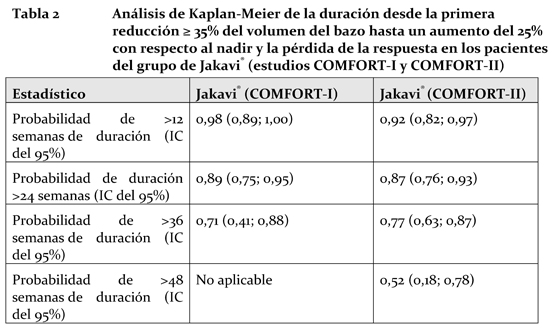
Entre los 80 pacientes que presentaron una reducción ≥35% en cualquier momento del estudio COMFORT-I y los 69 pacientes del estudio COMFORT-II, la probabilidad de que un paciente mantuviera una respuesta a Jakavi® durante por lo menos 24 semanas fue del 89% y del 87% en los estudios COMFORT-I y COMFORT-II, respectivamente, y la probabilidad de que se mantuviera una respuesta durante por lo menos 48 semanas fue del 52% en el estudio COMFORT-II. Jakavi® mejora los síntomas relacionados con la mielofibrosis y la calidad de vida de los pacientes con mielofibrosis (primaria o secundaria a policitemia vera o a trombocitemia idiopática). En el estudio COMFORT-I, los síntomas de mielofibrosis se registraron mediante el diario modificado (v. 2.0) del MFSAF (un diario electrónico que los sujetos completaban a diario). El cambio con respecto al inicio de la puntuación total a la semana 24 fue uno de los criterios secundarios de este estudio. Una proporción significativamente mayor de sujetos del grupo de Jakavi® presentó una mejora ≥50%, con respecto al inicio, de la puntuación total de síntomas a la semana 24 en comparación con el grupo del placebo (45,9% y 5,3%, respectivamente, p < 0,0001 usando la prueba de la ji al cuadrado). En ambos estudios COMFORT-I y COMFORT-II se apreció una mejora de la calidad de vida general al utilizar el cuestionario de calidad de vida (QLQ-C30) de la EORTC (Organización Europea para la Investigación y el Tratamiento del Cáncer). En el estudio COMFORT-I se comparó Jakavi® con el placebo al cabo de 24 semanas, y en el estudio COMFORT-II, Jakavi® con el mejor tratamiento disponible al cabo de 48 semanas. Al inicio de ambos estudios, las puntuaciones de las sub-escalas individuales del QLQ-C30 eran similares en los grupos de Jakavi® y de comparación. A la semana 24 del estudio COMFORT-I, según el QLQ-C30 de la EORTC, el grupo de Jakavi® presentó una mejora significativa de la calidad de vida o del estado de salud general en comparación con el grupo del placebo (cambio medio de +12,3 y -3,4 en los grupos de Jakavi® y del placebo, respectivamente, p < 0,0001). A las semanas 24 y 48, el grupo de Jakavi® del estudio COMFORT-II evidenció una tendencia hacia una mejora de la calidad de vida o del estado de salud general mayor que la lograda con el mejor tratamiento disponible -un criterio exploratorio-, lo cual concuerda con los resultados del estudio COMFORT-I. En el estudio COMFORT-I, fallecieron 10 de los 155 pacientes (6,5%) del grupo de Jakavi® y 14 de los 154 pacientes (9,1%) del grupo del placebo. En el estudio COMFORT-II, murieron 6 de los 146 pacientes (4,1%) del grupo de Jakavi® y 4 de los 73 pacientes (5,5%) del mejor tratamiento disponible. Propiedades Farmacocinéticas: Absorción: Ruxolitinib es una molécula de gran permeabilidad y solubilidad y rápida disolución que pertenece a la Clase 1 del Sistema de Clasificación de Productos Biofarmacéuticos. En los estudios clínicos, ruxolitinib se absorbió rápidamente después de la administración oral y alcanzó su concentración plasmática máxima (Cmáx) aproximadamente 1 hora después de la administración. Un estudio del balance de masas en seres humanos indicó que la absorción oral de ruxolitinib es del 95% o mayor. La Cmáx y la exposición total (ABC) medias del ruxolitinib aumentaron de forma proporcional a la dosis cuando se administraron dosis únicas de entre 5 y 200 mg. No se observaron alteraciones clínicamente significativas en la farmacocinética del ruxolitinib al administrar el medicamento con una comida cetógena. La Cmáx media disminuyó moderadamente (un 24%) pero el ABC media permaneció prácticamente invariable (aumentó un 4%) durante la administración con una comida cetógena. Distribución: En los pacientes con mielofibrosis, el volumen aparente de distribución en el estado estacionario es de 53-65 L. In vitro, casi el 97% del ruxolitinib presente en concentraciones de interés clínico se fija a proteínas plasmáticas, principalmente a la albúmina. Un estudio de distribución cuantitativa por autorradiografía del cuerpo entero efectuado en ratas reveló que ruxolitinib no atraviesa la barrera hematoencefálica. Biotransformación y metabolismo: Los estudios efectuados in vitro indican que la enzima CYP3A4 es la principal responsable del metabolismo del ruxolitinib. El compuesto original es la forma predominante en el ser humano y representa cerca del 60% de las sustancias circulantes vinculadas al fármaco. Se han identificado 2 metabolitos activos principales en el plasma de los sujetos sanos, que representan el 25% y el 11% del ABC original, respectivamente. Dichos metabolitos tienen entre la mitad y un quinto de la actividad farmacológica original relacionada con la JAK. Todos los metabolitos activos tomados en conjunto contribuyen al 18% de la farmacodinamia general del ruxolitinib. Los estudios in vitro indican que ruxolitinib, en concentraciones de interés clínico, no inhibe las enzimas CYP1A2, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6 o CYP3A4, ni es un inductor potente de las formas CYP1A2, CYP2B6 o CYP3A4. Eliminación: Tras la administración de una dosis oral única de [14C]-ruxolitinib a sujetos adultos sanos, la eliminación ocurrió predominantemente por vía metabólica, detectándose un 74% de radioactividad en la orina y un 22% en las heces. El fármaco inalterado representaba menos del 1% de la radioactividad total eliminada. La semivida de eliminación media del ruxolitinib es de 3 horas aproximadamente. Linealidad o no linealidad: La proporcionalidad a la dosis fue demostrada en los estudios de administración de dosis únicas y repetidas. Poblaciones especiales: Efectos de la edad, el sexo biológico o la raza: No se observaron diferencias significativas en la farmacocinética del ruxolitinib entre sujetos sanos de sexo o raza distintos. En un análisis farmacocinético de una población de pacientes con mielofibrosis, no se apreció una relación evidente entre la depuración oral y la edad o la raza de los pacientes. La depuración fue de 17,7 L/h en las mujeres y de 22,1 L/h en los varones y la variabilidad interindividual fue del 39%. Pacientes pediátricos: No se ha determinado la seguridad ni la eficacia de Jakavi® en los pacientes pediátricos. Insuficiencia renal: Después de la administración de una dosis única de 25 mg de ruxolitinib, la farmacocinética de los sujetos que padecían diversos grados de disfunción renal y de los sujetos con función renal normal fue similar. No obstante, las ABC plasmáticas de los metabolitos del ruxolitinib tendían a aumentar a medida que aumentaba la gravedad de la disfunción renal y de forma más pronunciada en los sujetos con nefropatía terminal que necesitaban hemodiálisis. Ruxolitinib no se elimina por diálisis. Se recomienda modificar la dosis en los pacientes con disfunción renal grave (depuración de creatinina menor que 30 mL/min). En los pacientes con nefropatía terminal se recomienda modificar la pauta posológica (ver Dosificación). Insuficiencia hepática: La farmacocinética y la farmacodinamia del ruxolitinib se evaluaron en sujetos con diversos grados de disfunción hepática que habían recibido una dosis única de 25 mg de ruxolitinib. El ABC media del ruxolitinib aumentó en un 87%, un 28% o un 65% en los pacientes con disfunción hepática leve, moderada o grave, respectivamente, en comparación con los pacientes con función hepática normal y no reveló ninguna relación clara con el grado de disfunción hepática a juzgar por las puntuaciones de Child-Pugh. En los pacientes con disfunción hepática, la semivida de eliminación terminal fue mayor que en los testigos sanos (4,1-5,0 horas frente a 2,8 horas). Se recomienda reducir la dosis en los pacientes con disfunción hepática (ver Dosificación). Datos de toxicidad preclínica: Ruxolitinib ha sido objeto de estudios de seguridad farmacológica, toxicidad tras dosis repetidas, genotoxicidad, toxicidad para la función reproductora y carcinogenia. Los órganos afectados asociados a la actividad farmacológica del ruxolitinib en los estudios de administración de dosis repetidas fueron la médula ósea, la sangre periférica y el tejido linfático. En los perros se apreciaron infecciones asociadas por lo general a la inmunodepresión. En un estudio de telemetría en perros se apreciaron reducciones de la tensión arterial y aumentos de la frecuencia cardíaca, y en otro estudio de la función respiratoria en ratas se observó una disminución del volumen minuto. Los límites (basados en la Cmáx del fármaco no unido a proteínas) con los no se produjeron efectos adversos en los estudios efectuados en perros y ratas fueron, respectivamente, 15,7 y 10,4 veces mayores que la máxima dosis humana recomendada (25 mg 2 veces al día). Una evaluación de los efectos neurofarmacológicos del ruxolitinib no reveló efecto alguno. El ruxolitinib no fue teratógeno, pero se asoció a un incremento de pérdidas post-implantacionales y a una disminución de los pesos fetales. No se observaron efectos sobre la fecundidad. En un estudio del desarrollo pre y post-natal, no se observaron efectos adversos en los índices de fecundidad ni en los parámetros de supervivencia, crecimiento y desarrollo embriofetales. Ruxolitinib no fue mutágeno ni clastógeno. Tampoco fue carcinógeno en el modelo de ratones transgénicos Tg.rasH2.
Indicaciones.
Jakavi® está indicado para el tratamiento de los pacientes con mielofibrosis, como la mielofibrosis primaria, la mielofibrosis secundaria a policitemia vera o la mielofibrosis secundaria a trombocitemia idiopática.
Dosificación.
Instrucciones para la supervisión: Hemogramas: antes de iniciar un tratamiento con Jakavi® debe realizarse un hemograma. Se supervisarán los hemogramas completos cada 2 o 4 semanas hasta que se estabilicen las dosis y, después, cuando esté indicado clínicamente (ver Advertencias y Precauciones). Dosis inicial: La dosis inicial recomendada de Jakavi® es de 15 mg administrados por vía oral, 2 veces al día, en los pacientes con cifras de trombocitos de entre 100000 y 200000/mm3, y de 20 mg 2 veces al día, en los pacientes con cifras de trombocitos > 200000/mm3. Se dispone de limitada información para recomendar una dosis inicial en los pacientes con cifras de trombocitos de entre 50000/mm3 y 100000/mm3. En dichos pacientes, se recomienda una dosis inicial máxima de 5 mg 2 veces al día, que luego debe ajustarse con cautela. Modificaciones posológicas: La dosis puede ajustarse en función de la seguridad y la eficacia. Debe interrumpirse el tratamiento si las cifras de trombocitos son menores que 50000/mm3 o las cifras absolutas de neutrófilos, inferiores a 500/mm3. En cuanto se hayan restablecido las cifras de trombocitos y de neutrófilos por encima de esos valores, el tratamiento puede reanudarse con 5 mg 2 veces al día, y luego se puede aumentar la dosis gradualmente basándose en la supervisión cuidadosa de los hemogramas. Si las cifras de trombocitos descienden por debajo de 100000/ mm3, considérese la posibilidad de reducir la dosis a fin de evitar la interrupción del tratamiento debido a trombocitopenia. Si la eficacia se considera insuficiente y las cifras de trombocitos y de neutrófilos son satisfactorias, la dosis puede incrementarse en 5 mg como máximo 2 veces al día. Durante las 4 primeras semanas de tratamiento no debe aumentarse la dosis inicial y pasado ese período puede aumentarse cada 2 semanas (nunca con mayor frecuencia). La dosis máxima de Jakavi® es de 25 mg 2 veces al día. Si se omite una dosis, el paciente no debe tomar una dosis adicional, sino la dosis usual siguiente tal como se le ha prescrito. El tratamiento puede continuar mientras proporcione más beneficios que riesgos al paciente. Ajuste de la dosis en caso de co-administración de inhibidores potentes del CYP3A4: Cuando Jakavi® se administra con inhibidores potentes del CYP3A4, la dosis diaria total de Jakavi® debe reducirse aproximadamente un 50%, disminuyendo ya sea la dosis diaria que se administra 2 veces al día o bien la frecuencia de administración a la correspondiente dosis de 1 vez al día si no resulta práctica la administración de 2 veces al día. Al instaurar un inhibidor potente del CYP3A4, se recomienda la supervisión más asidua de las magnitudes hemáticas y de los signos y síntomas clínicos de las reacciones adversas vinculadas a Jakavi®. Poblaciones especiales: Disfunción renal: En los pacientes con disfunción renal grave (depuración de creatinina inferior a 30 mL/min), la dosis inicial recomendada, que se basa en la cifra de trombocitos, debe reducirse un 50%. El paciente a quien se diagnostique una disfunción renal grave durante el tratamiento con Jakavi® debe ser objeto de una cuidadosa observación y puede que sea preciso reducir la dosis para evitar las reacciones adversas. Los datos de que se dispone para determinar las opciones posológicas óptimas para los pacientes con nefropatía terminal en diálisis son insuficientes. Los datos recabados en esta población indican que los pacientes en diálisis deben recibir una dosis única inicial de 15 o 20 mg, según la cifra de trombocitos, y dosis únicas ulteriores solamente después de cada sesión de diálisis y con una cuidadosa vigilancia de la seguridad y la eficacia. Disfunción hepática: En los pacientes con disfunción hepática, la dosis inicial recomendada, que se basa en la cifra de trombocitos, debe reducirse un 50%. Durante el tratamiento con Jakavi® se debe supervisar con cuidado a los pacientes con diagnóstico de disfunción hepática y puede que sea preciso reducir la dosis del mismo para evitar las reacciones adversas. Pacientes pediátricos: No se ha determinado la seguridad ni la eficacia de Jakavi® en los pacientes pediátricos. Pacientes geriátricos: No se recomienda efectuar ajustes adicionales de la dosis en los pacientes de edad avanzada. Modo de administración: Jakavi® se administra por vía oral, con o sin alimentos.
Contraindicaciones.
Hipersensibilidad a los principios activos o a cualquiera de los excipientes.
Reacciones adversas.
Resumen del perfil toxicológico: El perfil toxicológico de Jakavi® se ha evaluado en 617 pacientes tratados en 6 estudios en los que participaron sujetos con mielofibrosis, cáncer de próstata, mieloma múltiple, trombocitemia idiopática y policitemia vera. En el programa de estudios clínicos, se evaluó la gravedad de las reacciones adversas aplicando los Criterios Terminológicos Habituales para Acontecimientos Adversos (CTCAE), que definen los grados de gravedad (grado 1=leve, grado 2=moderado, grado 3=grave y grado 4=potencialmente mortal o incapacitante). En los 2 estudios fundamentales, COMFORT-I y COMFORT-II, la exposición mediana de 301 pacientes a Jakavi® fue de 9,6 meses (entre 2 semanas y 17 meses). La mayoría de los pacientes (el 55,8%) recibieron tratamiento durante 9 meses como mínimo. De los 301 pacientes expuestos, 111 (36,9%) presentaban una cifra inicial de trombocitos de entre 100000/mm3 y 200000/mm3, y 190 de ellos (63,1%), una cifra > 200000/mm3. Las reacciones adversas notificadas con más frecuencia fueron las náuseas y los vómitos. Entre las reacciones hemáticas (de cualquier grado CTCAE) figuran casos de anemia (81,7%), trombocitopenia (67,4%) y neutrocitopenia (15,3%). La anemia, la trombocitopenia y la neutrocitopenia son efectos relacionados con la dosis. Las 3 reacciones adversas no hemáticas más frecuentes fueron las equimosis (18,6%), los mareos (14,0%) y las cefaleas (12,6%). Las 3 anomalías analíticas no hemáticas más frecuentes fueron las elevaciones de alanina-aminotransferasa (26,2%) y de aspartato-aminotransferasa (18,6%) y la hipercolesterolemia (16,6%). En los estudios clínicos de Fase III, hubo que interrumpir el tratamiento debido a acontecimientos adversos en el 9,6% de los pacientes, con independencia de la causalidad. Resumen tabulado de reacciones adversas observadas en los ensayos clínicos: Las reacciones adversas descritas en los ensayos clínicos (Tabla 3) se han ordenado con arreglo a la clase de órgano, aparato o sistema del MedDRA. Dentro de cada clase de órgano, aparato o sistema, las reacciones adversas se clasifican por orden decreciente de frecuencia. También se indica la categoría de frecuencia de cada reacción adversa aplicando la siguiente convención (CIOMS III): muy frecuentes (≥1/10); frecuentes (≥1/100, < 1/10); poco frecuentes (≥1/1000, < 1/100); raras (≥1/10000, < 1/1000); muy raras ( < 1/10000).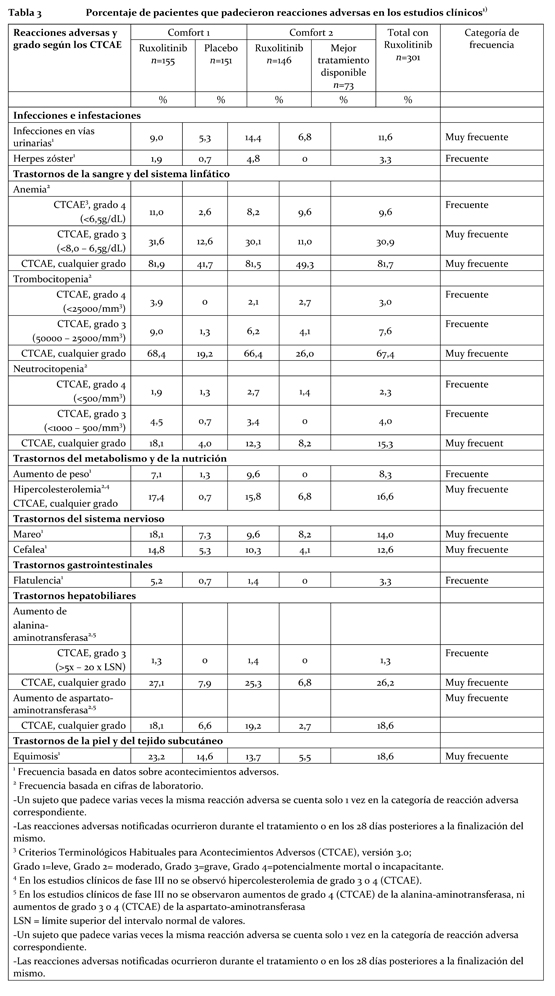
Al retirar el tratamiento, los pacientes pueden volver a padecer síntomas de mielofibrosis tales como cansancio, dolor óseo, fiebre, prurito, sudores nocturnos, esplenomegalia sintomática y pérdida de peso. En los estudios clínicos, la puntuación total de síntomas de mielofibrosis regresó paulatinamente a su nivel inicial en un plazo de 7 días después de la interrupción del tratamiento. Descripción de reacciones adversas específicas: Anemia: En los estudios clínicos de Fase III, la mediana del tiempo transcurrido hasta el inicio de la primera anemia de grado 2 o superior (CTCAE) fue de 1,5 meses. Un paciente (0,3%) abandonó el tratamiento debido a anemia. En los pacientes que recibieron Jakavi®, las disminuciones medias de hemoglobina alcanzaron un nadir de casi 15 a 20 g/L por debajo del valor inicial después de 8 a 12 semanas de tratamiento y luego se recuperaron gradualmente hasta alcanzar un nuevo estado estacionario de alrededor de 10 g/L por debajo del valor inicial. Esta pauta se observó con independencia de si el paciente había recibido transfusiones durante el tratamiento. En el estudio COMFORT-I, aleatorizado y comparativo con placebo, el 59,4% de los pacientes tratados con Jakavi® y el 37,1% de los pacientes del grupo del placebo recibieron transfusiones de eritrocitos durante el tratamiento aleatorizado. En el estudio COMFORT-II, la tasa de transfusiones de concentrado de eritrocitos fue del 51,4% en el grupo de Jakavi® y del 38,4% en el grupo que recibió el mejor tratamiento disponible. Trombocitopenia: En los pacientes que padecieron trombocitopenias de grado 3 o 4 en los estudios clínicos de fase III, la mediana del tiempo transcurrido hasta la aparición de dicho trastorno fue de aproximadamente 8 semanas. La trombocitopenia solía revertir al reducir la dosis o interrumpir la administración de la misma. La mediana del tiempo transcurrido hasta la recuperación de las cifras de trombocitos por encima de los 50000/mm3 fue de 14 días. Se hicieron transfusiones de trombocitos al 4,5% de los pacientes que recibieron Jakavi® y al 5,8% de los que recibieron los tratamientos de comparación. Se retiró el tratamiento debido a trombocitopenia en el 0,7% de los pacientes del grupo de Jakavi® y en el 0,9% de los pacientes que recibieron los tratamientos de comparación. Los pacientes con cifras de trombocitos de entre 100000/mm3 y 200000/mm3 antes de instaurar Jakavi® tuvieron una mayor frecuencia de trombocitopenias de grado 3 o 4 que los que tenían cifras de trombocitos > 200000/mm3 (64,2% frente a 35,4%). Neutrocitopenia: En los pacientes que padecieron neutrocitopenias de grado 3 o 4 en los estudios clínicos de Fase III, la mediana del tiempo transcurrido hasta la aparición de dicho trastorno fue de 12 semanas aproximadamente. Se comunicaron suspensiones o reducciones de la dosis debido a neutrocitopenias en el 1,3% de los pacientes, y el 0,3% de los pacientes interrumpieron el tratamiento a causa de ese trastorno. Infecciones en las vías urinarias: El 1,0% de los pacientes de los estudios clínicos de Fase III padecieron infecciones de grado 3 o 4 en las vías urinarias. Se notificó septicemia de origen urinario (urosepsis) en el 1,0% de los pacientes y una infección renal en 1 paciente. Herpes zóster: Se registró una infección por herpes zóster, de grado 3 o 4, en 1 paciente de los estudios clínicos de Fase III.
Precauciones.
Interacciones: Interacciones que obligan a reducir la dosis: Inhibidores potentes del CYP3A4: En los sujetos sanos que recibieron ketoconazol (un inhibidor potente del CYP3A4) en dosis de 200 mg 2 veces al día durante 4 días, el ABC de Jakavi® aumentó un 91% y la semivida se prolongó de 3,7 horas a 6,0 horas. Cuando Jakavi® se administra con inhibidores potentes del CYP3A4, su dosis diaria total debe reducirse un 50%. Es necesario vigilar estrechamente la aparición de citopenias y ajustar la dosis del paciente con arreglo a la seguridad y a la eficacia (ver Dosificación). Otras interacciones que se deben tomar en consideración: Inhibidores leves o moderados del CYP3A4: En los sujetos sanos que recibieron la eritromicina (un inhibidor moderado del CYP3A4) en dosis de 500 mg 2 veces al día durante 4 días, el ABC de Jakavi® aumentó un 27%. No se recomienda el ajuste de la dosis cuando Jakavi® se administre junto con inhibidores leves o moderados del CYP3A4 (como eritromicina). Es necesario vigilar estrechamente la aparición de citopenias cuando se inicie un tratamiento con un inhibidor moderado del CYP3A4. Inductores del CYP3A4: No se recomienda el ajuste de la dosis cuando se inicie un tratamiento con un inductor del CYP3A4. Cabe la posibilidad de aumentar gradualmente la dosis de Jakavi® si la efectividad terapéutica disminuye durante un tratamiento con dichos inductores. En los sujetos sanos que recibieron rifampicina (un inductor potente del CYP3A4) en dosis de 600 mg 1 vez al día durante 10 días, el ABC de Jakavi® (después de la administración de una dosis única) disminuyó un 71% y la semivida se redujo de 3,3 a 1,7 horas. Se apreció un aumento de la cantidad relativa de metabolitos activos con respecto al compuesto original. Glucoproteína P y otros transportadores: No se recomienda el ajuste de la dosis cuando Jakavi® se administra con sustancias que interactúan con la glucoproteína P y otros transportadores. Mujeres en edad de procrear: Las mujeres en edad de procrear deben tomar las precauciones necesarias para evitar el embarazo durante el tratamiento. En caso de embarazo, se deben sopesar los riesgos y los beneficios para la persona en cuestión y se debe brindar un cuidadoso asesoramiento sobre los riesgos que puede correr el feto usando los datos disponibles más recientes. Embarazo: No se han realizado estudios comparativos adecuados con Jakavi® en mujeres embarazadas. Los estudios de desarrollo embriofetal con ruxolitinib en ratas y conejos no arrojaron indicios de teratogenia. El ruxolitinib fue embriotóxico y fetotóxico en ratas (se registraron aumentos de pérdidas post-implantacionales y reducciones del peso fetal (ver Datos de toxicidad preclínica). Se desconoce el riesgo para el ser humano. No se recomienda el uso de Jakavi® durante la gestación. Lactancia: Las mujeres que toman Jakavi® no deben amamantar. Ruxolitinib y sus metabolitos se eliminan en la leche de ratas lactantes en una concentración 13 veces mayor que la concentración plasmática materna. No se sabe si ruxolitinib pasa a la leche humana. Fecundidad: No se dispone de datos sobre los efectos del ruxolitinib en la fecundidad humana. En los estudios en animales, no se observaron efectos sobre la fecundidad o la función reproductora de las ratas machos o hembras. En un estudio pre y post-natal en ratas, tampoco se observó afectación de la fecundidad de las crías de la primera generación. Efectos sobre la capacidad de conducir vehículos y utilizar máquinas: No se han estudiado los efectos sobre la capacidad de conducir vehículos y utilizar máquinas.
Advertencias.
Disminución de células sanguíneas: El tratamiento con Jakavi® puede provocar reacciones hemáticas adversas tales como trombocitopenia, anemia y neutrocitopenia. Antes de comenzar el tratamiento con Jakavi® debe realizarse un hemograma completo (ver Dosificación). Se ha observado que los pacientes con cifras reducidas de trombocitos ( < 200000/mm3) al inicio del tratamiento son más propensos a padecer trombocitopenias durante la terapia. El tratamiento de la trombocitopenia suele ser reversible y consiste usualmente en la reducción de la dosis o la interrupción temporal de Jakavi®. No obstante, pueden necesitarse transfusiones de trombocitos, si están indicadas clínicamente (ver Dosificación). Los pacientes que padezcan anemias pueden necesitar transfusiones sanguíneas. En dichos pacientes también debe considerarse la posibilidad de modificar la dosis. La neutrocitopenia (cifra absoluta de neutrófilos [CAN] < 500/mm3) suele ser reversible y su tratamiento consiste en la interrupción temporal de Jakavi® (ver Dosificación y Reacciones adversas). Se deben vigilar los hemogramas completos cuando sea clínicamente conveniente y se debe ajustar la dosis si fuera necesario (ver Dosificación y Reacciones adversas). Infecciones: Se debe evaluar el riesgo de que el paciente padezca bacteriosis, micobacteriosis, micosis