LIPOMAX 35
LIPOMAX 105
BAGO
Normolipemiante. Activador del subtipo alfa del receptor activado por proliferador de peroxisomas (PPAR-alfa, por sus siglas en inglés).
Composición.
Cada Comprimido de Lipomax 35 contiene: Ácido Fenofíbrico 35,00 mg. Excipientes: Copovidona 3,00 mg, Povidona Reticulada 2,00 mg, Estearato de Magnesio 0,50 mg, Celulosa Microcristalina c.s.p. 100,00 mg. Cada Comprimido de Lipomax 105 contiene: Ácido Fenofíbrico 105,00 mg. Excipientes: Copovidona 9,00 mg, Povidona Reticulada 6,00 mg, Estearato de Magnesio 1,50 mg, Celulosa Microcristalina c.s.p. 300,00 mg.
Farmacología.
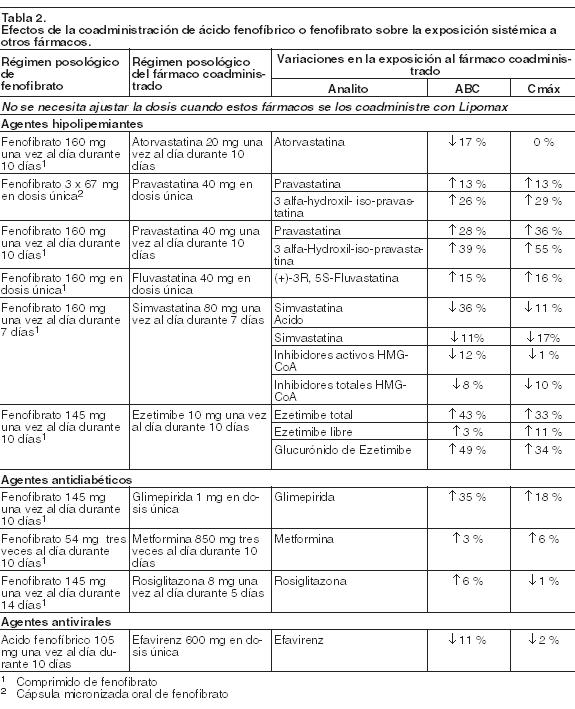
Acción farmacológica: Las propiedades hipolipemiantes del Ácido Fenofíbrico observadas en la práctica clínica han sido explicadas in vivo en los ratones transgénicos y sobre cultivos de hepatocitos humanos por la activación del receptor PPAR- alfa. Gracias a este mecanismo, el Ácido Fenofíbrico aumenta la lipólisis y la eliminación del plasma de las partículas ricas en TG mediante la activación de la lipoproteinlipasa y la reducción de la producción de apoproteína C-III (la cual ejerce un efecto inhibidor sobre la lipoproteinlipasa). La consiguiente disminución de los TG produce una alteración en el tamaño y composición del C-LDL, transformándolo de partículas pequeñas y densas (que se suponen aterógenas por su suceptibilidad a la oxidación) a partículas grandes y flotantes. Estas partículas más grandes tienen mayor afinidad por los receptores del colesterol y son catabolizadas rápidamente. La activación de los PPAR-alfa también induce un incremento en la síntesis de C-HDL y de las apoproteínas A-I y A-II (Apo A-I y A-II). Los niveles elevados de C-total, C-LDL y Apo B y los niveles disminuidos de C-HDL y de su complejo de transporte, la Apo A-I y la Apo A-II, son factores de riesgo para la aterosclerosis humana. Los estudios epidemiológicos han establecido que la morbimortalidad cardiovascular varía en forma directamente proporcional a los niveles de C-total, C-LDL y TG, y en forma inversamente proporcional a los niveles de C-HDL. El efecto independiente de elevar el C-HDL o de reducir los TG sobre el riesgo de morbimortalidad cardiovascular no ha sido determinado. Farmacocinética: Absorción: Tras la administración oral de Ácido Fenofíbrico en voluntarios sanos, la media de las concentraciones plasmáticas máximas de Ácido Fenofíbrico ocurre aproximadamente 2,5 horas luego de su administración. El Ácido Fenofíbrico se puede ingerir con o sin alimentos, dado que su influencia sobre la absorción no es clínicamente significativa. El alcance y la velocidad de absorción de Ácido Fenofíbrico después de la administración de 105 mg son equivalentes a la administración de 145 mg de fenofibrato en ayunas. Distribución: Tras la administración repetida, el estado de equilibrio constante del Ácido Fenofíbrico se logra en un plazo de 9 días. Las concentraciones plasmáticas de Ácido Fenofíbrico en estado de equilibrio son un poco más del doble de las que siguen a una dosis única. La unión a proteínas séricas es aproximadamente del 99 % en sujetos normales y en pacientes con diagnóstico de hiperlipidemia. Metabolismo: El Ácido Fenofíbrico se conjuga principalmente con ácido glucurónico y luego se excreta a través de la orina. Una pequeña cantidad de Ácido Fenofíbrico se reduce a nivel de la fracción carbonilo a un metabolito benzidrol el que, a su vez, se conjuga con ácido glucurónico y se excreta a través de la orina. Excreción: La excreción se realiza esencialmente por vía urinaria en forma de Ácido Fenofíbrico y su conjugado glucurónico. Se elimina con una vida media de 20 horas, permitiendo la administración de Lipomax una vez al día. Poblaciones especiales: Uso en ancianos: En 5 voluntarios de edad avanzada, de 77 a 87 años de edad, el aclaramiento oral de Ácido Fenofíbrico después de una dosis oral única de fenofibrato fue de 1,2 l/h, frente a 1,1 l/h en los adultos jóvenes. En pacientes ancianos con función renal normal se pueden utilizar dosis equivalentes a las administradas a pacientes adultos jóvenes, sin que se produzca acumulación del fármaco o de sus metabolitos. Uso en pediatría: No se ha investigado el uso de Lipomax en ensayos clínicos controlados en esta población. Sexo: No se observaron diferencias farmacocinéticas entre hombres y mujeres con el uso de fenofibrato. Raza: La influencia de la raza sobre la farmacocinética de Ácido Fenofíbrico no ha sido estudiada. Sin embargo, el Ácido Fenofíbrico no es metabolizado por las enzimas actualmente conocidas que presentan variabilidad racial. Insuficiencia hepática: No hay estudios efectuados de la farmacocinética del Ácido Fenofíbrico en pacientes con insuficiencia hepática. Insuficiencia renal: La farmacocinética del Ácido Fenofíbrico fue estudiada en pacientes con insuficiencia renal leve, moderada y severa. Los pacientes con insuficiencia renal severa (clearance de creatinina ≤ 30 ml/min) exhibieron un aumento de 2,7 veces en la exposición al Ácido Fenofíbrico y un aumento en la acumulación de Ácido Fenofíbrico durante la administración prolongada en comparación con sujetos sanos. Los pacientes con insuficiencia renal leve (clearance de creatinina 50-80 ml/min) a moderada (clearance de creatinina 30-50 ml/min) tuvieron una exposición similar, pero registraron un incremento en la vida media del Ácido Fenofíbrico en comparación con la de sujetos sanos. Basándose en estos hallazgos, el uso de Lipomax se deberá evitar en pacientes que presentan insuficiencia renal severa, y reducir la dosis en pacientes con insuficiencia renal leve a moderada. Interacciones farmacológicas: Estudios in vitro que utilizaron hepatocitos humanos, indican que el Ácido Fenofíbrico no induce al CYP1A2, CYP2A6, CYP2B6, CYP2C9, CYP2C19, CYP2D6, CYP2E1 y CYP3A4. Estudios in vitro que emplearon microsomas de hígado humano indican que el Ácido Fenofíbrico no es un inhibidor de las isoformas del CYP450: CYP3A4, CYP2D6, CYP2E1 o CYP1A2. La Tabla 1 describe los efectos de los fármacos coadministrados sobre la exposición sistémica de Ácido Fenofíbrico. La Tabla 2 describe los efectos del Ácido Fenofíbrico coadministrado sobre la exposición con otros fármacos.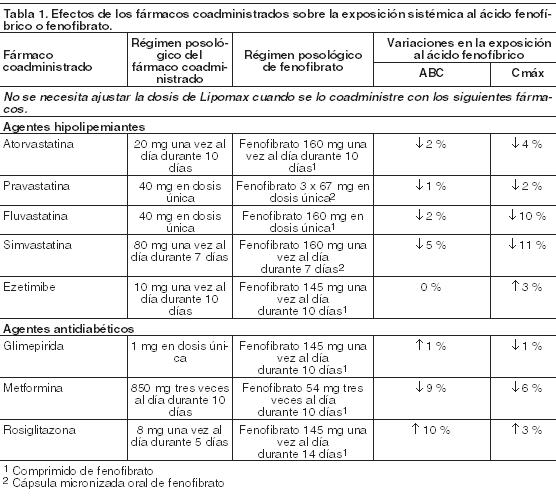
Estudios clínicos: Tratamiento concomitante con estatinas: Se evaluó la eficacia y seguridad del Ácido Fenofíbrico coadministrado con estatinas en tres estudios clínicos publicados doble ciego y controlados de fase 3, de 12 semanas de duración y un estudio de extensión a largo plazo, abierto, de 52 semanas de duración, en 2698 pacientes con dislipidemia mixta. Los pacientes debían cumplimentar los siguientes criterios lipídicos en ayunas al ingreso: TG > 150 mg/dl, c-hdl < 40 mg/dl (varones) y < 50 mg/dl (mujeres) y C-LDL > 130 mg/dl. Los tres estudios multicéntricos doble-ciego, aleatorizados y controlados poseían diseños similares y diferían principalmente en la estatina empleada para el tratamiento combinado / monoterapia. Estos estudios compararon los efectos sobre los factores de riesgo lipídicos de cardiopatía coronaria de 135 mg de Ácido Fenofíbrico coadministrado con una dosis baja o una dosis moderada de estatina vs Ácido Fenofíbrico en monoterapia y la estatina en monoterapia en dosis correspondientes. Un grupo más pequeño de pacientes recibió una dosis alta de estatina en monoterapia. En el estudio 1, los pacientes recibieron Ácido Fenofíbrico coadministrado con 10 mg o 20 mg de rosuvastatina. En el estudio 2, los pacientes recibieron Ácido Fenofíbrico coadministrado con 20 mg o 40 mg de simvastatina. En el estudio 3, los pacientes recibieron Ácido Fenofíbrico coadministrado con 20 mg o 40 mg de atorvastatina. Los pacientes fueron incorporados durante un total de aproximadamente 22 semanas, consistente en un período basal (libre) de fármacos, un período de tratamiento de 12 semanas y un período de seguimiento de 30 días por razones de seguridad. Los pacientes que habían completado el período de tratamiento de 12 semanas podían ingresar al estudio de extensión a largo plazo de 52 semanas. De los 2698 pacientes aleatorizados y tratados en los estudios controlados, el 51,6% eran mujeres y el 48,4% varones; el 92,6% de todos los sujetos era de raza blanca, el 4,7% de raza negra y el 2,8 % de otras razas. Los pacientes de origen latino comprendían el 9,9 % de la población del estudio. La edad media fue de 54,9 años. Los principales criterios de valoración de la eficacia de los tres estudios fueron las variaciones porcentuales medias desde los valores basales a los valores finales en el C-HDL, TG y C-LDL. Con cada dosis de estatina coadministrada con Ácido Fenofíbrico se hicieron tres comparaciones principales. Para el C-HDL y TG, el Ácido Fenofíbrico coadministrado con cada dosis de estatina se comparó con la estatina administrada en monoterapia en la dosis correspondiente. Para el C-LDL, el Ácido Fenofíbrico coadministrado con cada dosis de estatina se comparó con Ácido Fenofíbrico administrado en monoterapia. Para declarar que el tratamiento de combinación resultó exitoso para una dosis de estatina en particular, las tres comparaciones principales debían demostrar la superioridad del tratamiento combinado sobre la monoterapia correspondiente. Los principales resultados de la eficacia fueron similares en los tres estudios y confirmados por el análisis combinado de los tres estudios. Los resultados de cada estudio en particular y del análisis combinado demostraron que el Ácido Fenofíbrico coadministrado con estatinas en dosis bajas y en dosis moderadas fue superior a la monoterapia correspondiente. Se observaron diferencias estadísticamente significativas en las tres comparaciones principales de la eficacia para ambas dosis de tratamiento combinado en los tres estudios doble-ciego y controlados, así como en el análisis combinado. En el análisis combinado, la coadministración de Ácido Fenofíbrico con estatinas tanto en dosis bajas como en dosis moderadas derivó en incrementos porcentuales medios (18,1 % y 17,5 %) en el C-HDL y en reducciones porcentuales medias (-43,9% y -42,0 %) en los TG significativamente superiores que con la dosis correspondiente de estatina en monoterapia (7,4 % y 8,7 % para el C-HDL; -16,8 % y -23,7% para los TG). Además ambas dosis del tratamiento combinado provocó disminuciones porcentuales medias (-33,1 % y -34,6 %) en el C-LDL significativamente superiores que con la monoterapia con Ácido Fenofíbrico (-5,1 %). Un total de 1895 pacientes que completaron las 12 semanas de tratamiento en los estudios doble-ciego y controlados fueron tratados en el estudio de extensión a largo plazo de 52 semanas. Los pacientes recibieron Ácido Fenofíbrico coadministrado con la dosis moderada de estatina que había sido empleada en el estudio doble-ciego y controlado en el que estaban participando. Ya sea que el tratamiento combinado se hubiera iniciado durante los estudios doble-ciego y controlados, o durante el estudio de extensión a largo plazo, el efecto terapéutico del tratamiento combinado se observó dentro de las cuatro semanas, y se sostuvo durante todo el estudio a largo plazo. Un total de 568 pacientes completó las 52 semanas de tratamiento con Ácido Fenofíbrico coadministrado con estatinas. Los valores medios de las 52 semanas y la variación porcentual media desde los valores basales (al momento de la incorporación a los estudios aleatorizados y controlados) fueron de 91,7 mg/dl (-38,2%) para el C-LDL, 47,3 mg/dl (+24,0%) para el C-HDL, 135 mg/dl (-47,6%) para los TG, 117,9 mg/dl (-45,7%) para el C-NoHDL, 26,2 mg/dl (-53,1 %) para el C-VLDL, 165,2 mg/dl (-35,4 %) para el C-total y 81,4 mg/dl (-43,6 %) para la Apo B. Hipertrigliceridemia: En dos estudios clínicos publicados doble-ciego, aleatorizados y controlados contra placebo, que incorporaron 147 pacientes hipertrigliceridémicos, se estudiaron los efectos del fenofibrato sobre los TG séricos. Los pacientes fueron tratados durante 8 semanas bajo protocolos que diferían únicamente en que uno ingresó pacientes con niveles basales de TG de 350 a 500 mg/dl. En los pacientes con hipertrigliceridemia y colesterolemia normal con o sin hiperquilomicronemia, el tratamiento con fenofibrato en dosis equivalentes a 135 mg una vez al día de Ácido Fenofíbrico redujo principalmente los TG-VLDL y el C-VLDL. El tratamiento de pacientes con TG elevados a menudo aumenta el C-LDL. Hipercolesterolemia primaria (familiar heterocigótica y no familiar) y dislipidemia mixta: Los efectos del fenofibrato en una dosis equivalente a 135 mg de Ácido Fenofíbrico una vez al día se evaluaron en cuatro estudios publicados doble-ciego, aleatorizados, controlados contra placebo, de grupos paralelos, que incorporaron pacientes con los siguientes valores basales medios de lípidos: C-Total de 306,9 mg/dl, C-LDL de 213,8 mg/dl, C-HDL de 52,3 mg/dl y TG de 191,0 mg/dl. El tratameinto con fenofibrato redujo el C-LDL, el C-Total y la relación C-LDL/C-HDL. La terapéutica con fenofibrato también redujo los TG y elevó el C-HDL. En un grupo de sujetos, se practicaron determinaciones de la Apo B. El tratamiento con fenofibrato redujo significativamente la Apo B desde el valor basal hasta el criterio de valoración en comparación con el placebo (-25,1 % vs 2,4 %, p < 0,0001, n=213 y 143 respectivamente).
Indicaciones.
Tratamiento de la hipertrigliceridemia severa: Está indicado como complemento de la dieta para reducir los niveles de triglicéridos (TG) en pacientes con hipertrigliceridemia severa (≥ 500 mg/dl). Los niveles extremadamente elevados de TG séricos (por ej. > 1000 mg/dl) pueden aumentar el riesgo de desarrollar pancreatitis. El efecto de la intervención terapéutica con Ácido Fenofíbrico en la reducción de este riesgo, aún no ha sido estudiado en forma adecuada. Un mejor control glucémico en pacientes diabéticos que presentan quilomicronemia en ayunas obviará la necesidad de una intervención farmacológica. Tratamiento de la hiperlipidemia primaria o de la dislipidemia mixta: Está indicado como complemento de la dieta para reducir los niveles elevados de colesterol total (C-total), colesterol LDL (C-LDL), apoproteína B y TG y para aumentar el colesterol HDL (C-HDL), en pacientes con diagnóstico de hipercolesterolemia primaria o dislipidemia mixta. Consideraciones generales para el tratamiento: Lipomax contiene como principio activo Ácido Fenofíbrico, cuyos efectos farmacológicos han sido ampliamente estudiados a través de la administración oral de fenofibrato, que es convertido in vivo en Ácido Fenofíbrico.
Dosificación.
Es indispensable el cumplimiento de la dieta por parte del paciente antes y durante el tratamiento con el medicamento. Lipomax puede darse independientemente de las comidas. Se debe confirmar que los niveles de lípidos son anormales previo a instituir el tratamiento con Lipomax. Se deben efectuar todos los esfuerzos para controlar el nivel de los lípidos séricos con dieta apropiada, ejercicio, descenso de peso en pacientes obesos y el control de ciertas patologías como la diabetes mellitus y el hipotiroidismo, que contribuyen de manera variable a las anormalidades en los lípidos. Los medicamentos que exacerban la hipertrigliceridemia (beta-bloqueantes, diuréticos tiazídicos, estrógenos) deben suspenderse o reemplazarse antes del inicio del tratamiento con Lipomax. Deben obtenerse determinaciones periódicas de lípidos en suero durante el inicio de la terapia con el fin de establecer la dosis mínima eficaz de Lipomax. El tratamiento debe discontinuarse en pacientes que no obtienen una respuesta adecuada luego de dos meses de tratamiento con la dosis máxima recomendada de 105 mg por día. Se debería considerar la posibilidad de reducir la dosis de Lipomax si los niveles de lípidos caen significativamente por debajo del rango objetivo. Dosis recomendada en adultos: Hipertrigliceridemia severa: la dosis inicial de Lipomax es de 35 a 105 mg/día. La dosis debe adecuarse según la respuesta de cada paciente debiendo ser ajustada, de ser necesario, siguiendo a determinaciones lipídicas seriadas, con intervalos de 4-8 semanas. Hiperlipidemia primaria o dislipidemia mixta: la dosis de Lipomax es de 105 mg/día. Pacientes con insuficiencia renal: en pacientes con insuficiencia renal leve a moderada, el tratamiento con Lipomax debe iniciarse con una dosis de 35 mg/día, y sólo aumentarse después de evaluar los efectos sobre la función renal y sobre los niveles lipídicos con esta dosis. El uso de Lipomax debe evitarse en pacientes con insuficiencia renal grave (ver Farmacología - Farmacocinética, Poblaciones especiales). Ancianos: la selección de dosis para las personas de edad avanzada debe adecuarse a la función renal (ver Farmacología - Farmacocinética, Poblaciones especiales).
Contraindicaciones.
Lipomax está contraindicado en: Enfermedad hepática activa, incluyendo cirrosis biliar primaria o anomalías persistentes inexplicables de la función hepática. Enfermedad pre-existente de la vesícula biliar. Hipersensibilidad conocida al Ácido Fenofíbrico, al fenofibrato o a cualquier otro componente de la formulación (ver Advertencias). Insuficiencia renal grave, incluidos aquellos que reciben diálisis. Madres durante el período de lactancia.
Reacciones adversas.
Experiencia en ensayos clínicos: Dado que los ensayos clínicos se llevan a cabo en condiciones muy diversas, las tasas de reacciones adversas observadas en los ensayos clínicos de un fármaco no pueden compararse directamente con las tasas en los ensayos clínicos de otro fármaco y pueden no reflejar las tasas observadas en la práctica clínica. Las reacciones adversas notificadas en un 2 % o más de los pacientes tratados con fenofibrato (y mayor que con placebo) durante el ensayo doble ciego, controlados con placebo, se muestran en la Tabla 3. Las reacciones adversas llevaron a la interrupción del tratamiento en el 5 % de los pacientes tratados con fenofibrato y en el 3 % de los tratados con placebo. El aumento de las enzimas hepáticas fue el evento más frecuente, causando la interrupción del tratamiento con fenofibrato en el 1,6 % de los pacientes.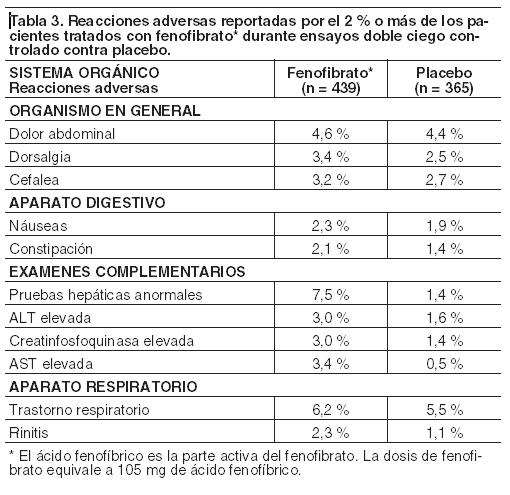
Experiencia poscomercialización: Las siguientes reacciones adversas han sido identificadas durante el uso posterior a la aprobación de fenofibrato. Debido a que estas reacciones son reportadas voluntariamente por una población de tamaño incierto, no siempre es posible estimar de manera fiable su frecuencia o establecer una relación causal con la exposición al fármaco. Se han reportado: mialgia, rabdomiólisis, aumento de creatinfosfoquinasa, pancreatitis, aumento de transaminasas hepáticas, espasmo muscular, insuficiencia renal aguda, hepatitis, cirrosis, náuseas, dolor abdominal, anemia, cefalea, artralgias y astenia.
Advertencias.
Pruebas de función hepática anormal: El Ácido Fenofíbrico y el fenofibrato (administrado en un rango de dosis, con la mayor equivalente a 105 mg de Ácido Fenofíbrico) se ha asociado con un aumento de las transaminasas en suero (AST o ALT). En un análisis conjunto de 10 ensayos clínicos controlados con placebo, se evidenció un aumento de más de 3 veces el límite superior del rango normal de ALT en el 5,3 % de los pacientes que tomaban fenofibrato frente al 1,1% de los pacientes tratados con placebo. La incidencia de los aumentos de las transaminasas observados con el tratamiento con fenofibrato podría estar relacionada con la dosis. Han sido reportadas hepatitis crónica activa y hepatitis colestásica asociada al tratamiento con fenofibrato después de la exposición de semanas a varios años. En casos extremadamente raros, la cirrosis ha sido reportada en asociación con la hepatitis crónica activa. El monitoreo periódico de las enzimas hepáticas (por ejemplo, ALT) se debe realizar durante la terapia con Lipomax debiendo suspender el mismo si los niveles de la enzima persisten elevados más de tres veces el límite superior normal. Colelitiasis: Lipomax, como el fenofibrato, clofibrato y gemfibrozil, puede aumentar la excreción de colesterol en la bilis y producir así colelitiasis. Si se sospecha colelitiasis, se deben efectuar estudios de la vesícula biliar. La terapia con Lipomax debe suspenderse ante la presencia de cálculos biliares. Uso concomitante con anticoagulantes orales: Se debe tener precaución cuando Lipomax se da conjuntamente con anticoagulantes orales. Lipomax puede potenciar los efectos anticoagulantes de estos fármacos resultando en la prolongación del tiempo de protrombina/Razón Internacional Normalizada (RIN). La monitorización frecuente del tiempo de protrombina/RIN y el ajuste de la dosis del anticoagulante se recomienda hasta que el tiempo de protrombina / RIN se ha estabilizado con el fin de evitar complicaciones hemorrágicas. Miopatía: Los fibratos incrementan el riesgo de miopatía y se han asociado con rabdomiólisis. El riesgo de toxicidad muscular grave parece ser mayor en pacientes ancianos y en pacientes con diabetes, insuficiencia renal o hipotiroidismo. Los datos de estudios observacionales sugieren que el riesgo de rabdomiólisis aumenta cuando los fibratos, en particular gemfibrozil, se administran conjuntamente con un inhibidor de la HMG-CoA reductasa (estatinas). El diagnóstico de miopatía debe considerarse en cualquier paciente que presente mialgias difusas, sensibilidad o debilidad muscular y/o elevaciones marcadas de los niveles de creatinfosfoquinasa (CPK). Los pacientes deben ser advertidos de informar sin demora, si presentan dolor, sensibilidad o debilidad muscular inexplicable, especialmente si son acompañados por malestar o fiebre. Los niveles de CPK deben ser evaluados en pacientes que presentaron estos síntomas y la terapia con Lipomax debe suspenderse si los niveles de CPK son marcadamente elevados o si se diagnostica miopatía. Elevación de creatinina sérica: Elevaciones en el valor de la creatinina sérica se han notificado en pacientes tratados con fenofibrato. Estos aumentos tienden a regresar a los valores basales tras la interrupción de fenofibrato. La importancia clínica de estas observaciones se desconoce. El seguimiento de la función renal debe considerarse en pacientes con diagnóstico o riesgo de padecer insuficiencia renal, como los ancianos y los pacientes diabéticos. Morbilidad y mortalidad cardiovascular: El efecto de Lipomax sobre la morbimortalidad por cardiopatía coronaria y la morbimortalidad no cardiovascular no ha sido establecido. Debido a las similitudes existentes entre Lipomax y el fenofibrato, el clofibrato y el gemfibrozil, los hallazgos de los siguientes estudios clínicos aleatorizados y controlados contra placebo, llevados a cabo en gran escala con estos fibratos, también podrían aplicarse a Lipomax. El estudio Intervención con Fibratos y la Reducción de Eventos en Pacientes Diabéticos (FIELD) fue un estudio aleatorizado, controlado con placebo, con una duración de 5 años, que incorporó 9775 pacientes con diabetes mellitus tipo 2 tratados con fenofibratos. El fenofibrato demostró una reducción no significativa del 11 % en el criterio principal de valoración de eventos coronarios, (Riesgo Relativo (RR) 0,89; IC 95%: 0,75-1,05; p=0,16) y una reducción significativa del 11 % en el criterio de valoración secundario de eventos cardiovasculares totales (RR 0,89; 0,80-0,99; p=0,04). Se observó un incremento no significativo del 11 % (RR 1,11; 0,95 a 1,29; p=0,18) y del 19% (RR 1,19; 0,90, 1,57; p=0,22) en la mortalidad total y por cardiopatía coronaria, respectivamente, con fenofibrato en comparación con el placebo. El Proyecto de Medicamentos para la Enfermedad Coronaria, un estudio publicado que incorporó pacientes post-infarto de miocardio tratados durante 5 años con el clofibrato, no reveló diferencias en la mortalidad, entre el grupo tratado con clofibrato y el grupo que recibió placebo. Sin embargo, hubo una diferencia en la incidencia de casos de colelitiasis y colecistitis que requirieron cirugía entre los dos grupos (3,0 % vs 1,8 %). En un estudio realizado por la Organización Mundial de la Salud (OMS), 5000 sujetos sin enfermedad coronaria conocida fueron tratados con placebo o clofibrato durante 5 años y seguidos por un período adicional de un año. Se observó una mortalidad por cualquier causa ajustada por edad, superior y estadísticamente significativa en el grupo que recibió clofibrato en comparación con el grupo que recibió placebo (5,70% vs 3,96%, p < 0,01). El exceso de mortalidad se debió a un aumento del 33% en las causas no cardiovasculares, incluyendo tumores malignos, complicaciones post-colecistectomía y pancreatitis. Esto parece confirmar el mayor riesgo de desarrollar enfermedad de la vesícula biliar en los pacientes tratados con clofibrato estudiados en el Proyecto de Medicamentos para la Enfermedad Coronaria. El Estudio de Corazón de Helsinki fue realizado sobre una muestra significativa (n=4081) de hombres de mediana edad sin una historia de enfermedad coronaria. Los sujetos recibieron placebo o gemfibrozil durante 5 años, con un período de extensión abierto posterior de 3,5 años. La mortalidad total fue numéricamente más alta en el grupo aleatorizado a gemfibrozil, pero no alcanzó significación estadística (p = 0,19, IC =0,91-1,64). Si bien la mortalidad por cáncer tendió a ser superior en el grupo que recibió gemfibrozil (p = 0,11), las neoplasias (excluido el carcinoma de basocelular) se diagnosticaron con igual frecuencia en ambos grupos de tratamiento. Debido al tamaño limitado del estudio, el riesgo relativo de muerte por cualquier causa no demostró ser diferente al derivado de los datos de seguimiento de 9 años del estudio de la OMS (RR=1,29). Asimismo el exceso numérico de cirugías de vesícula en el grupo gemfibrozil no se diferenció estadísticamente de lo observado en el estudio de la OMS. Un componente de prevención secundaria del Estudio de Corazón de Helsinki incorporó hombres de mediana edad excluidos del estudio de prevención primario debido a cardiopatía coronaria conocida o sospechada. Los sujetos recibieron gemfibrozil o placebo durante 5 años. Si bien la mortalidad cardíaca fue superior en el grupo tratado con gemfibrozil, esto no revistió significación estadística (RR de 2,2; IC del 95 %: 0,94-5,05). El promedio de cirugías de vesícula no fue estadísticamente significativa entre los grupos de estudio, pero tuvo una tendencia más elevada en el grupo tratado con gemfibrozil (1,9 % vs 0,3 %, p=0,07). Hubo una diferencia estadísticamente significativa en el número de apendicectomías en el grupo tratado con gemfibrozil (6/311 vs 0/317, p= 0,029). Pancreatitis: La pancreatitis se ha informado en pacientes tratados con fenofibrato. Este hecho podría representar una falta de eficacia en pacientes con hipertrigliceridemia grave, un efecto directo del fármaco, o un fenómeno secundario mediado a través de cálculos biliares o la formación de barro biliar con obstrucción del conducto biliar común. Enfermedad tromboembólica: En el Estudio FIELD, la embolia pulmonar (EP) y la trombosis venosa profunda (TVP) se observaron en mayor proporción con el fenofibrato que en el grupo tratado con placebo. De 9775 pacientes incluidos en el estudio, hubo 4900 en el grupo placebo y 4875 en el grupo de fenofibrato. Para la TVP, se registraron 48 eventos (1 %) en el grupo placebo y 67 (1%) en el grupo de fenofibrato (p=0,074), y para la EP, hubo 32 (0,7 %) eventos en el grupo placebo y 53 (1 %) en el grupo de fenofibrato (p=0,022). En el Proyecto de Medicamentos para la Enfermedad Coronaria, una mayor proporción del grupo tratado con clofibrato experimentó episodios confirmados o presentes de embolia pulmonar fatal o no fatal y/o tromboflebitis en comparación al grupo que recibió placebo (5,2 % vs 3,3 % a cinco años, p < 0,01). Reacciones de hipersensibilidad: Se han reportado reacciones agudas de hipersensibilidad en los individuos tratados con fenofibrato, incluyendo erupciones cutáneas severas poco frecuentes como el síndrome de Stevens-Johnson y la necrólisis epidérmica tóxica, que requirieron la hospitalización del paciente y tratamiento con esteroides. Cambios hematológicos: Se ha observado disminución en la concentración de hemoglobina, de leve a moderada, en el hematócrito y en el recuento de glóbulos blancos tras el inicio del tratamiento con fenofibrato. La trombocitopenia y la agranulocitosis han sido reportados en los individuos tratados con fenofibrato. El control periódico con hemogramas seriados se recomienda durante los primeros 12 meses de la administración de Lipomax. Interacción con otros medicamentos y otras formas de tratamiento: Anticoagulantes orales: se debe tener precaución cuando Lipomax se administra en combinación con anticoagulantes cumarínicos. Lipomax puede potenciar el efecto anticoagulante de estos fármacos, prolongando el tiempo de protrombina/RIN. Se recomienda el monitoreo frecuente del tiempo de protrombina/RIN con el ajuste consiguiente de la dosis del anticoagulante oral, hasta que el tiempo de protrombina se haya estabilizado a fin de evitar complicaciones hemorrágicas (ver Advertencias). Resinas fijadoras de ácidos biliares: dado que los fármacos secuestradores de ácidos biliares pueden unirse a otros agentes que se administren en forma concomitante, los pacientes deben ingerir Lipomax por lo menos 1 hora antes o 4-6 horas después de tomar esta clase de fármacos para evitar que interfiera con su absorción. Inmunosupresores: los agentes inmunosupresores como la ciclosporina y el tacrolimus pueden deteriorar la función renal. Cuando estos inmunosupresores y otros fármacos potencialmente nefrotóxicos se administran conjuntamente con Lipomax, la menor dosis efectiva de este último debe ser empleada y la función renal debe ser controlada. Carcinogénesis, mutagénesis, deterioro de la fertilidad: Carcinogénesis: Dos estudios de carcinogénesis han sido conducidos en ratas con fenofibrato en la dieta. En el primer estudio de 24 meses de duración, ratas Wistar recibieron fenofibrato en dosis de 10, 45 y 200 mg/kg/día (aproximadamente 0, 3, 1, y 6 veces la dosis máxima recomendada para seres humanos (DMRH) en base al área de superficie corporal (mg/m2). En una dosis de 200 mg/kg/día (6 veces la DMRH), la incidencia de carcinomas hepáticos fue significativamente aumentada en ambos sexos. Se observó un incremento estadísticamente significativo de carcinomas pancreáticos en los machos con dosis de 45 y 200 mg/kg/día (aproximadamente 1 y 6 veces la DMRH), un aumento de adenomas pancreáticos y tumores testiculares benignos de células intersticiales se registró en machos en una dosis 6 veces superiores a la DMRH. En un segundo estudio de 24 meses de duración de carcinogénesis en una variedad diferente de ratas (Sprague-Dawley), las dosis de 10 y 60 mg/kg/día (1,2 y 16,5 veces la DMRH de Ácido Fenofíbrico, basado sobre la exposición) produjeron aumentos significativos en la incidencia de adenomas acinares pancreáticos en ambos sexos y de incrementos en tumores testiculares de células intersticiales en los machos con dosis 16,5 veces la DMRH de fenofibrato (60 mg/kg/día). Se llevó a cabo un estudio de carcinogénesis de 117 semanas de duración en ratas que comparó tres fármacos: fenofibrato 10 y 60 mg/kg/día (0, 3 y 2 veces la DMRH de fenofibrato), clofibrato (400 mg/kg/día; 2 veces la dosis humana), y gemfibrozil (250 mg/kg/día; 2 veces la dosis humana) en base a mg/m2 de superficie corporal. El fenofibrato aumentó la incidencia de adenomas acinares pancreáticos en ambos sexos. El clofibrato incrementó el carcinoma hepatocelular y los adenomas acinares pancreáticos en los machos y nódulos neoplásicos hepáticos en las hembras. El gemfibrozil aumentó los nódulos neoplásicos hepáticos en machos y hembras, mientras que los tres fármacos incrementaron los tumores testiculares de células intersticiales en los machos. En un estudio de 21 meses de duración en ratones CF-1, el fenofibrato administrado en dosis de 10, 45 y 200 mg/kg/día (aproximadamente 0,2; 0,7 y 3 veces la DMRH en base a mg/m2 de superficie corporal) aumentó considerablemente los carcinomas hepáticos en ambos sexos en las dosis que resultan de la exposición al Ácido Fenofíbrico que es 3,7 veces más la DMRH de Ácido Fenofíbrico. En un segundo estudio de 18 meses de duración en dosis de 10, 60 y 200 mg/kg/día, el fenofibrato aumentó significativamente los carcinomas hepáticos en ratones machos y adenomas hepáticos en ratones hembras con dosis 3 veces mayor a la DMRH de fenofibrato. Los estudios que emplearon técnicas de microscopía de electrones han demostrado la proliferación peroxisomal después de la administración de fenofibrato a ratas. No se ha llevado a cabo un estudio adecuado para examinar la proliferación de peroxisomas en seres humanos, pero se han observado variaciones en la morfología y cantidad de peroxisomas en humanos tras el tratamiento con otros miembros de la clase de los fibratos cuando se compararon biopsias hepáticas anteriores y posteriores al tratamiento en el mismo individuo. Mutagénesis: El fenofibrato ha demostrado estar exento de potencial mutágeno en los siguientes ensayos: Ames, linfoma de ratón, aberraciones cromosómicas y síntesis de ADN no programada en hepatocitos primarios de ratas e in vivo en el ensayo de micronúcleo de ratón. Deterioro de la fertilidad: En estudios de fertilidad, se le administró a ratas dosis orales de fenofibrato en la dieta. Los machos recibieron las dosis durante 61 días antes del apareamiento y las hembras desde 15 días antes del apareamiento hasta el destete, lo que no causó ningún efecto adverso sobre la fertilidad en dosis de hasta 300 mg/kg/día (aproximadamente 10 veces más la DMRH de fenofibrato, en base a comparaciones en mg/m2 de superficie corporal). Embarazo: categoría C: La seguridad en mujeres embarazadas no ha sido establecida. No hay estudios adecuados y bien controlados de Ácido Fenofíbrico en mujeres embarazadas. El Ácido Fenofíbrico debe utilizarse durante el embarazo sólo si el beneficio potencial justifica el riesgo potencial para el feto. En ratas hembras que recibieron dosis orales de 15, 75 y 300 mg/kg/día de fenofibrato desde 15 días antes del apareamiento hasta el destete, se observó toxicidad materna en una exposición al Ácido Fenofíbrico que es aproximadamente 50 veces más que en la DMRH de Ácido Fenofíbrico. En ratas preñadas que recibieron dosis orales en la dieta de 14, 127 y 361 mg/kg/día desde el día 6-15 de gestación durante el período de organogénesis no se observaron trastornos del desarrollo con 14 mg/kg/día (en una exposición al Ácido Fenofíbrico que es aproximadamente el doble que en la DMRH de Ácido Fenofíbrico). Fue observada toxicidad materna con múltiplos más altos de las dosis para seres humanos. En conejas preñadas que recibieron dosis orales por sonda nasogástrica de 15, 150 y 300 mg/kg/día desde el día 6-18 de gestación, durante el período de la organogénesis, y a las que se les permitió parir, se registraron camadas abortadas con la dosis de 150 mg/kg/día (20 veces mayor que la DMRH de Ácido Fenofíbrico). No se observaron anomalías congénitas con la dosis de 15 mg/kg/día (7 veces mayor que en la MRHD de Ácido Fenofíbrico). En ratas preñadas que recibieron dosis orales en la dieta de 15, 75 y 300 mg/kg/día a partir del día 15 de gestación hasta el día 21 de lactancia (destete), se observó toxicidad materna en una exposición al Ácido Fenofíbrico que es aproximadamente 50 veces más que en la DMRH de Ácido Fenofíbrico. Lactancia: Lipomax no debe ser usado durante la lactancia (ver Contraindicaciones). Uso pediátrico: La seguridad y efectividad de Lipomax en pacientes pediátricos no han sido establecidas. Uso geriátrico: Lipomax se excreta principalmente por el riñón como Ácido Fenofíbrico y como glucurónico de Ácido Fenofíbrico y el riesgo de reacciones adversas de este fármaco puede ser mayor en pacientes con deterioro de la función renal. Dado que los ancianos presentan una alta incidencia de insuficiencia renal, la elección de la dosis para este grupo etario debe basarse en su función renal (ver Farmacología, Farmacocinética, Poblaciones especiales y Dosificación). Insuficiencia renal: El uso de Lipomax debe ser evitado en pacientes con severo deterioro de la función renal. En pacientes con insuficiencia renal leve a moderada se requiere reducción de la dosis. Se recomienda el monitoreo de la función renal en pacientes con insuficiencia renal (ver Farmacología, Farmacocinética, Poblaciones especiales y Dosificación). Insuficiencia hepática: El uso de Lipomax no ha sido evaluado en sujetos con disfunción hepática (ver Contraindicaciones).
Conservación.
Conservar el producto a una temperatura no mayor de 30 °C. Mantener en su envase original.
Sobredosificación.
No existe un tratamiento específico para la sobredosis con Lipomax. Debe indicarse cuidados generales de soporte del paciente, incluyendo el monitoreo de signos vitales y observación del estado clínico. Si está indicado, la eliminación del fármaco no absorbido puede lograrse mediante emesis o lavado gástrico y observarse las precauciones habituales para mantener la permeabilidad de la vías aéreas. Debido a que Lipomax se une ampliamente a las proteínas plasmáticas, la hemodiálisis no debe ser considerada. Ante la eventualidad de una sobredosificación concurrir al Hospital más cercano o comunicarse con los Centros de Toxicología: Hospital de Niños Ricardo Gutiérrez, Tel.: (011) 4962-6666/2247, Hospital Pedro de Elizalde (ex Casa Cuna), Tel.: (011) 4300-2115, Hospital Nacional Prof. Dr. Alejandro Posadas, Tel.: (011) 4654-6648/4658-7777.
Presentación.
Lipomax 35: Envases conteniendo 30 comprimidos de color blanco con puntos amarillos. Lipomax 105: Envases conteniendo 30 comprimidos color blanco con puntos amarillos.