HEMAX®
BIOSIDUS S.A.U
Antianémico. Estimulante de la eritropoyesis.
Descripción.
HEMAX es un medicamento que contiene Eritropoyetina humana recombinante (r-HuEPO, epoetina) como principio activo. La eritropoyetina es una glicoproteína de 165 aminoácidos producida por técnicas de recombinación de ADN, obtenida a partir de una línea celular de mamífero genéticamente modificada. La r-HuEPO posee un nivel máximo de pureza y es indistinguible de la Eritropoyetina humana natural.
Composición.
Cada vial con polvo liofilizado contiene: HEMAX® 1000 UI/ml: Eritropoyetina humana Recombinante 1000 UI. Excipientes: Manitol 25,0 mg. Cloruro de sodio 3,2 mg. Fosfato monobásico de sodio 1,4 mg. Fosfato dibásico de sodio dodecahidratado 4,0 mg. Albúmina humana 2,5 mg. HEMAX® 2000 UI/2ml: Eritropoyetina humana Recombinante 2000 UI. Excipientes: Manitol 50,0 mg. Cloruro de sodio 6,4 mg. Fosfato monobásico de sodio 2,8 mg. Fosfato dibásico de sodio dodecahidratado 8,0 mg. Albúmina humana 5,0 mg. HEMAX® 3000 UI/2ml: Eritropoyetina humana Recombinante 3000 UI. Excipientes: Manitol 50,0 mg. Cloruro de sodio 6,4 mg. Fosfato monobásico de sodio 2,8 mg. Fosfato dibásico de sodio dodecahidratado 8,0 mg. Albúmina humana 5,0 mg. Cada ampolla con diluyente contiene: HEMAX®1000 UI/ml: Agua para inyectables 1 ml. Cada ampolla con diluyente contiene: HEMAX®2000 UI/2ml: Agua para inyectables 2 ml. HEMAX®3000 UI/2ml: Agua para inyectables 2 ml. HEMAX®4000 UI/2ml: Agua para inyectables 2 ml. HEMAX®10000 UI/ml: Agua para inyectables 1 ml. HEMAX®20000 UI/ml: Agua para inyectables 1 ml. HEMAX®40000 UI/ml: Agua para inyectables 1 ml. Cada vial con solución contiene: Hemax® 2000 UI/ml: Eritropoyetina Humana Recombinante 2000 UI. Excipientes: Manitol 25,0 mg. Cloruro de sodio 3,2 mg. Fosfato monobásico de sodio 1,4 mg. Fosfato dibásico de sodio dodecahidratado 4,0 mg. Albúmina humana 2,5 mg. Agua para inyectables 1 ml. HEMAX® 4000 UI/ml: Eritropoyetina Humana Recombinante 4000 UI. Excipientes: Manitol 25,0 mg. Cloruro de sodio 3,2 mg. Fosfato monobásico de sodio 1,4 mg. Fosfato dibásico de sodio dodecahidratado 4,0 mg. Albúmina humana 2,5 mg. Agua para inyectables 1 ml.
Farmacología.
A) Mecanismo de Acción: La eritropoyetina induce la eritropoyesis al estimular la división y diferenciación de progenitores eritropoyéticos en médula ósea, lo que resulta en un aumento de la masa globular y, en consecuencia, del hematócrito. La eritropoyetina también induce la liberación de reticulocitos desde médula ósea hacia torrente sanguíneo, donde maduran a eritrocitos. La concentración normal de eritropoyetina endógena es de 10-30 mU/ml y está influenciada por los niveles de oxígeno a nivel tisular. Cuando los niveles de oxígeno tisular disminuyen, la concentración de eritropoyetina aumenta entre 100 y 1.000 veces. Esta situación también se observa en los pacientes anémicos. B) Farmacocinética: La Epoetina, principio activo de HEMAX®, se administra por vía parenteral (subcutánea o intravenosa). El incremento inicial en el recuento de reticulocitos se produce dentro de los 7 a 10 días posteriores a la administración. Se observan incrementos clínicamente significativos en el recuento de glóbulos rojos, hematócrito y hemoglobina, generalmente, en las 2 a 6 semanas posteriores a la administración de epoetina. El rango y extensión de la respuesta depende de la dosis y disponibilidad de las reservas de hierro. La máxima concentración plasmática se logra luego de 15 minutos tras una dosis única intravenosa y entre las 5 a 24 horas posteriores a la administración por vía subcutánea de una única dosis. En este último caso, las concentraciones pico pueden mantenerse por 12 a 16 horas y presentar cantidades detectables durante por lo menos 24 horas luego de su administración. La vida media de la epoetina alfa es de 4 a 13 horas tras su administración intravenosa o subcutánea. La vida media de eliminación es generalmente mayor luego de las primeras dosis que luego de dos o más semanas de tratamiento. En general, transcurridas 24 horas los niveles plasmáticos de eritropoyetina regresan a su nivel basal. Luego de la administración subcutánea de epoetina, la concentración máxima de la droga se observa entre las 5 y 24 horas posteriores a la administración, y su declinación es más lenta. En estudios efectuados sobre voluntarios adultos sanos, se observó que la vida media luego de la administración endovenosa es un 20 % menor que en los pacientes con insuficiencia renal. En un estudio efectuado en voluntarios sanos, se observó que la vida media de HEMAX® administrada por vía subcutánea fue de 20,8 ± 6,3 horas. Una vez interrumpido el tratamiento, el hematócrito puede comenzar a disminuir luego de 2 semanas.
Indicaciones.
HEMAX® está indicado para: Tratamiento de la anemia asociada a la insuficiencia renal crónica: HEMAX® está indicado en pacientes adultos y pediátricos, tanto en aquéllos que están en diálisis como en los que no requieren diálisis, con el objetivo de elevar o mantener el nivel de glóbulos rojos (determinado por los valores de hematócrito o hemoglobina) y reducir la necesidad de transfusiones. Sin embargo, los pacientes con anemia sintomática que no se encuentran en diálisis deberán tener una hemoglobina inferior a 10 g/dl para ser considerados aptos para el tratamiento con HEMAX®. HEMAX® no debe ser utilizado como sustituto de una transfusión de emergencia en pacientes que requieran una corrección inmediata de una anemia severa. Tratamiento de la anemia en pacientes infectados por VIH tratados con zidovudina: HEMAX® está indicado para el tratamiento de la anemia asociada al tratamiento con zidovudina en pacientes infectados con el VIH, con el objeto de elevar o mantener el nivel de glóbulos rojos (determinado por los valores de hematócrito o hemoglobina) y reducir la necesidad de transfusiones. No está indicado para el tratamiento de la anemia vinculada a otros factores (déficit de hierro o folatos, hemólisis, hemorragia gastrointestinal) en este grupo de pacientes. Tratamiento de la anemia en pacientes con cáncer que reciben quimioterapia: En pacientes con neoplasias metastásicas no mieloides, HEMAX® está indicado para el tratamiento de la anemia sintomática ocasionada por la administración de quimioterapia. El tratamiento con epoetina ha demostrado reducir la necesidad de transfusiones de glóbulos rojos en pacientes que recibían quimioterapia concomitante durante un período mínimo de 2 meses. HEMAX® no está indicado para el tratamiento de la anemia vinculada a otros factores (déficit de hierro o folatos, hemólisis, hemorragia gastrointestinal) en este grupo de pacientes. HEMAX® no está indicado en pacientes que reciben tratamiento con productos hormonales, biológicos o radioterapia sin quimioterapia mielosupresora concomitante. No está indicado el uso de HEMAX® en pacientes con tratamiento quimioterápico en los cuales se prevea curación. Reducción del número de transfusiones alogénicas en pacientes anémicos sometidos a cirugías programadas: HEMAX® está indicado en pacientes anémicos (hemoglobina mayor a 10 y menor o igual a 13 g/dl), con alto riesgo de presentar hemorragias perioperatorias, sometidos a cirugías programadas, no cardíacas ni vasculares, para reducir la necesidad de transfusiones sanguíneas alogénicas. Está indicado para pacientes con alto riesgo de necesitar transfusiones perioperatorias, por una probabilidad anticipada de una significativa pérdida de sangre. No está indicado para pacientes anémicos que serán donantes autólogos. Tratamiento de la anemia del prematuro: HEMAX® está indicado para el tratamiento de la anemia del prematuro en recién nacidos pretérmino con peso de nacimiento entre 750 - 1.500 g y edad gestacional menor a 34 semanas. HEMAX® no debe ser utilizado como sustituto de una transfusión de emergencia en pacientes que requieren una corrección inmediata de una anemia severa.
Dosificación.
A) Tratamiento de la anemia secundaria a insuficiencia renal crónica: La insuficiencia renal crónica es un cuadro clínico en el que se observa una progresiva disminución de la función renal en forma irreversible. El tratamiento con epoetina ha demostrado estimular la eritropoyesis en los pacientes con anemia e insuficiencia renal, requieran o no diálisis. La primera evidencia de la estimulación de la eritropoyesis es el aumento de los reticulocitos a los 8 días de haber iniciado el tratamiento; posteriormente, entre la 2ª y 6ª semana, se observa el aumento de la hemoglobina y el hematócrito. La velocidad y magnitud de este aumento dependen de la dosis inicial de epoetina alfa, los niveles basales de hematócrito y hemoglobina, las reservas de hierro y las situaciones clínicas que pueden provocar resistencia al tratamiento (estados inflamatorios, infecciosos, etc.). Antes del inicio del tratamiento con HEMAX® deben descartarse otras causas de anemia (ej., déficit de ácido fólico o vitamina B12) y corregir factores concomitantes que puedan agravar la anemia, en especial el déficit de hierro. Por lo tanto, deben realizarse estudios del metabolismo del hierro que incluyan ferremia, capacidad total de saturación y porcentaje de saturación de la transferrina, y ferritina sérica. Se recomienda que los pacientes tengan una saturación de transferrina mayor al 20 % y más de 100 ng/dl de ferritina antes de iniciar el tratamiento con HEMAX®. Los niveles de hierro deben ser monitoreados y mantenidos en valores adecuados durante el tratamiento con epoetina. La tensión arterial debe controlarse previo al tratamiento, y monitorearse estrictamente durante el mismo. La dosis inicial recomendada en pacientes adultos en hemodiálisis es de 50 UI/Kg/dosis por vía i.v. o de 40 UI/Kg/dosis vía s.c., tres veces por semana. Luego de cuatro semanas de tratamiento, la dosis debe modificarse de acuerdo al aumento alcanzado en los niveles de hemoglobina: Si el aumento es de 1 g/dl o mayor: continuar con la misma dosis. Si el aumento es inferior a 1 g/dl: aumentar la dosis en incrementos de 25 UI/Kg/dosis. La dosis máxima sugerida es de 300 UI/Kg tres veces por semana. Una vez alcanzado el valor deseado, se puede reducir la dosis en un 30% y pasar a la vía s.c. si es que el paciente había iniciado tratamiento por vía i.v. La dosis de mantenimiento debe ser individualizada para cada paciente. El 10% de los pacientes en diálisis requiere 25 UI/Kg/dosis tres veces por semana, y otro 10% requiere 200 UI/Kg/dosis tres veces por semana; la dosis promedio de mantenimiento es de 75 UI/Kg/dosis tres veces por semana. Los ajustes de dosis deben efectuarse a intervalos no menores a 4 semanas ya que la respuesta al cambio de dosis se evidencia luego de 2 a 6 semanas. Los pacientes con insuficiencia renal que no requieren diálisis responden al tratamiento de igual manera que los que están en diálisis. Las dosis recomendadas están entre 75 y 100 UI/Kg/semana; se recomienda usar la vía s.c. En pacientes pediátricos la dosis inicial recomendada es igual a la de los adultos. La dosis de mantenimiento depende del tamaño corporal. Las dosis usadas habitualmente, en administración tres veces por semana, son: a) peso inferior a 10 kg: 75 a 150 UI/Kg/dosis; b) peso entre 10 y 30 kg: 60 a 150 UI/Kg/dosis; c) peso superior a 30 kg: 30 a 100 UI/Kg/dosis. La dosis debe reducirse gradualmente hasta el valor más bajo que permita mantener hematócrito y hemoglobina en los niveles deseados. B) Pacientes infectados por el virus VIH tratados con zidovudina: HEMAX® reduce el requerimiento transfusional y aumenta el hematócrito en los pacientes VIH positivos tratados con zidovudina, dando como resultado un aumento significativo en la calidad de vida. Los pacientes con niveles de eritropoyetina endógena inferiores a 500 mU/ml responden mejor al tratamiento, por lo que se recomienda realizar un dosaje de eritropoyetina previo al mismo. La dosis inicial recomendada es de 100 UI/Kg/dosis en adultos y 150 UI/Kg/dosis en niños, tres veces por semana, por vía i.v o s.c durante 8 semanas. La respuesta puede evaluarse a las 4 semanas de tratamiento. En caso de no obtenerse una respuesta satisfactoria, esta dosis se puede incrementar de a 50 UI/Kg hasta un máximo de 300 UI/Kg tres veces por semana. La respuesta al tratamiento con epoetina alfa puede disminuir en presencia de infecciones o episodios inflamatorios. Si los valores de hematócrito exceden el 40%, la administración de HEMAX® puede interrumpirse hasta que dicho valor llegue a 36%. La dosis debe reducirse en un 25% cuando se reanude el tratamiento y luego evaluar si el hematócrito se mantiene en los valores deseados. C) Anemia en pacientes con cáncer que reciben quimioterapia: En esta población de pacientes, la epoetina aumenta el hematócrito y reduce las necesidades transfusionales entre el 1ro y el 4to mes de tratamiento. Existen dos regímenes de administración de HEMAX® que pueden emplearse: a) Administración tres veces por semana: La dosis inicial recomendada es de 150 UI/Kg/dosis tres veces por semana por vía s.c. En caso de no obtenerse respuesta a las 8 semanas, la dosis se puede aumentar de a 50 UI/Kg/dosis hasta un máximo de 300 UI/Kg/dosis tres veces por semana. Si la hemoglobina llega a 12 g/dl o aumenta más de 1 g/dl en un período de 2 semanas, reducir la dosis en un 25%. Si los valores de hematócrito exceden el 40%, la administración puede interrumpirse hasta que dicho valor llegue a 36%. La dosis debe reducirse en un 25% cuando se reanude el tratamiento y luego evaluar si el hematócrito se mantiene en los valores deseados. En el caso de pacientes pediátricos cuyas edades oscilan entre los 6 meses y 18 años, las dosis reportadas fueron de 25 a 300 UI/Kg i.v o s.c, tres a siete veces por semana. b) Administración de dosis única semanal: La dosis inicial en adultos es de 40.000 UI, vía s.c., una vez por semana. Si luego de 4 semanas la hemoglobina no aumentó 1 g/dl, en ausencia de transfusión, la dosis de HEMAX® debe aumentarse a 60.000 UI. Si el tratamiento con HEMAX® produce una respuesta rápida, por ejemplo un aumento de hemoglobina mayor de 1 g/dl en un período de 2 semanas, la dosis debe reducirse en un 25%. La administración de HEMAX® debe interrumpirse si el valor de hemoglobina excede 13 g/dl, y reiniciarse con una dosis reducida en un 25% cuando la hemoglobina haya descendido a un nivel inferior a 12 g/dl. Se recomienda discontinuar el tratamiento aproximadamente a las 4 semanas de haber terminado la quimioterapia. Si los pacientes no han respondido satisfactoriamente a la dosis semanal de 60.000 UI luego de 4 semanas, es poco probable que respondan a dosis mayores de HEMAX®. En pacientes pediátricos se han utilizado dosis semanales de 10.000 a 20.000 UI. D) Transfusión autóloga: En pacientes con cirugía electiva (cadera, rodilla, etc.) en programa de autotransfusión se demostró que la utilización de epoetina alfa permite reducir la necesidad de transfusiones alogénicas. La principal variable predictiva de respuesta al tratamiento es el valor de hemoglobina previo a la cirugía; los pacientes con niveles entre 10 y 13 g/dl son los que más se benefician con esta terapia. La dosis inicial es de 300 UI/Kg/día por vía s.c., comenzando 10 días antes de la cirugía y continuando hasta 4 días posteriores a la misma. Como esquema alternativo, puede administrarse en dosis únicas semanales de 600 UI/Kg vía s.c., los días 21, 14 y 7, previos a la cirugía y la cuarta dosis el día de la cirugía. Todos los pacientes deben recibir un suplemento de hierro adecuado, cuya administración deberá iniciarse a más tardar al comienzo del tratamiento con HEMAX® y continuar durante todo el curso del mismo. E) Anemia del prematuro: El uso de HEMAX® en la anemia del prematuro reduce los requerimientos transfusionales, medidos tanto en el número de pacientes transfundidos como en el volumen de sangre transfundido. La dosis recomendada es de 250 UI/Kg tres veces por semana, por vía s.c., a partir de la segunda semana de vida y durante ocho semanas.
Contraindicaciones.
HEMAX® está contraindicado en pacientes con: 1. Hipertensión arterial no controlada. 2. Aplasia pura de serie roja luego de tratamiento previo con epoetina. 3. Hipersensibilidad conocida a la albúmina humana. 4. Hipersensibilidad conocida a productos derivados de líneas celulares de mamíferos.
Reacciones adversas.
Pacientes con insuficiencia renal crónica: a) Hipertensión arterial: Más del 80 % de los pacientes en hemodiálisis tienen antecedentes de hipertensión arterial. Cuando se inicia el tratamiento con epoetina alfa debe controlarse estrictamente la tensión arterial de los pacientes y adecuarse los tratamientos antihipertensivos así como también las restricciones alimentarias. El 25 % de los pacientes que reciben epoetina sufren un aumento de la tensión arterial, requiriendo ajustes en la medicación antihipertensiva. Existe una probable relación entre la velocidad de aumento del hematócrito y la exacerbación de la tensión arterial, por lo que se recomienda reducir la dosis de HEMAX® si el hematócrito aumenta más de 4 % en un período de 2 semanas. b) Aplasia pura de células rojas: Dado que la epoetina es una proteína, algunos pacientes pueden sintetizar anticuerpos contra HEMAX®. Algunos casos de aplasia pura de la serie roja se han asociado con anticuerpos neutralizantes contra productos que contenían epoetina alfa. Todos los enfermos padecían de insuficiencia renal y recibían la droga por vía subcutánea. Dichos pacientes no podrán recibir HEMAX® ni ningún otro producto que contenga epoetina. c) Eventos trombóticos: Se ha reportado un aumento de fenómenos trombóticos en pacientes en diálisis con patología cardiovascular concomitante que recibieron epoetina alfa. Éstos consistieron en trombosis de la fístula arterio-venosa, infarto agudo de miocardio y otros. Los eventos trombóticos se observaron en pacientes asignados a alcanzar valores de hematócrito mayores de 40 %. Asimismo, este grupo tuvo aumento de la mortalidad. Durante la diálisis los pacientes pueden requerir un aumento de la dosis de heparina para prevenir la trombosis de la fístula. Los niveles de hemoglobina mayores que 12 g/dl pueden ser asociados con un riesgo mayor de eventos cardiovasculares. d) Convulsiones: En ensayos clínicos con epoetina alfa se observó que 2,5 % de pacientes adultos en diálisis tratados sufrieron convulsiones, generalmente asociadas a crisis de hipertensión arterial. Debe controlarse estrictamente la tensión arterial antes y durante el tratamiento. La epoetina alfa deberá administrarse con precaución en pacientes con antecedentes de episodios convulsivos. Pacientes infectados con VIH tratados con zidovudina: A diferencia de los pacientes con insuficiencia renal, en este grupo no se ha reportado exacerbación de la hipertensión arterial, convulsiones, ni eventos trombóticos. Pacientes con cáncer tratados con quimioterapia: Se ha observado una mayor incidencia de episodios trombóticos e incremento de mortalidad en pacientes con cáncer de mama que recibían quimioterapia, asignadas a tratamiento con epoetina alfa con el objetivo de mantener niveles elevados de hemoglobina (12 a 14 g/dl). Albúmina (humana): HEMAX® contiene albúmina, un derivado de la sangre humana. El riesgo de transmisión de enfermedades virales es extremadamente remoto, basado en los procesos de obtención de la albúmina y de manufactura del producto. El riesgo teórico de transmisión de enfermedad de Creutzfeldt-Jakob también es considerado remoto. No han sido identificados casos de transmisión de enfermedades virales por albúmina. La tabla que sigue enumera las reacciones adversas que suelen requerir atención médica: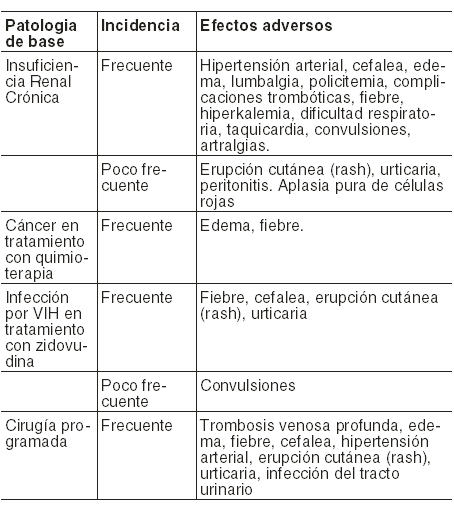
En la tabla siguiente se presentan las reacciones adversas que suelen requerir atención médica sólo en caso de que se prolonguen en el tiempo o interfieran en la actividad diaria:
Precauciones.
Inmunogenicidad: Al igual que para todo producto que se administre por vía parenteral, debe tenerse precaución por eventuales reacciones alérgicas que pudieran manifestarse luego de la administración de HEMAX®. En los estudios clínicos se han reportado reacciones alérgicas menores y transitorias. No se observaron reacciones anafilácticas o reacciones alérgicas serias con el uso de epoetina alfa. Hematología: Se ha comunicado exacerbación de la porfiria en pacientes en diálisis tratados con epoetina alfa. Si bien este evento no se observa con frecuencia, deberá tenerse en cuenta en pacientes con antecedentes de porfiria. Pérdida o disminución de la respuesta: En pacientes que estén recibiendo dosis de mantenimiento y manifiesten disminución o pérdida de la respuesta a la epoetina alfa deben descartarse las siguientes causas: 1. Déficit de hierro, 2. Infección, inflamación o neoplasia, 3. Pérdida de sangre oculta por materia fecal, 4. Disfunción de la médula ósea por patología hematológica asociada (talasemia, mielodisplasia, etc.), 5. Hemólisis, 6. Intoxicación por aluminio, 7. Déficit de vitamina B12 o ácido fólico, 8. Fibrosis quística, 9. Aplasia pura de la serie roja. Suplemento de hierro: Los requerimientos de hierro pueden incrementarse si los depósitos de hierro existentes fueron empleados para la eritropoyesis y en algunos pacientes puede ser necesario un suplemento de hierro. En algunos pacientes, la suplementación de hierro por vía oral puede ser insuficiente y requerir la administración de hierro sacarato por vía intravenosa. Interacciones con otras drogas: No hay evidencias de interacciones entre HEMAX® y otras drogas. Carcinogénesis y mutagénesis: El potencial carcinogénico de HEMAX® no ha sido evaluado. La epoetina alfa no induce mutaciones genéticas en bacterias ni aberraciones cromosómicas en células de mamíferos. Fertilidad: En ratas tratadas con dosis de 100 a 500 UI/kg de epoetina alfa por vía endovenosa, se observó una tendencia a un leve aumento de abortos. Embarazo: - FDA Categoría C. No hay suficientes estudios sobre el uso de HEMAX® durante el embarazo. Por tal motivo este producto sólo debe usarse en caso que se juzgue que el beneficio a obtener justifique el potencial riesgo para el feto. En ratas preñadas se observó aumento de las pérdidas fetales. En conejos tratados con dosis de 500 UI/Kg no se observó ningún efecto adverso. Lactancia: La eritropoyetina humana es un componente normal de la leche, aunque su papel en la misma no está claro. Se desconoce si HEMAX® es excretado en la leche materna. Debido a que muchas drogas se excretan por leche materna, debe tenerse precaución en las mujeres que estén amamantando si reciben HEMAX®. Uso Pediátrico: Si bien se han realizado múltiples estudios en recién nacidos, lactantes y niños mayores, demostrando que HEMAX® es efectivo y seguro para la prevención y el tratamiento de la anemia, no se ha establecido aún la seguridad de este producto a largo plazo. Controles de laboratorio: Luego de iniciado el tratamiento, deberá controlarse la hemoglobina o el hematócrito 2 veces por semana hasta alcanzar el valor buscado (10 a 12 g/dl, o 30 a 36 %, respectivamente). Una vez alcanzado ese valor deberán efectuarse controles semanales, durante cuatro semanas, para determinar si el mismo se mantiene estable. Posteriormente la determinación se hará a intervalos regulares. Los recuentos de plaquetas, glóbulos blancos y glóbulos rojos y la determinación de la concentración de hemoglobina deberán realizarse en forma regular (cada 4 semanas). Se ha descripto moderado aumento del número de plaquetas en pacientes tratados con HEMAX®, no significativos desde el punto de vista clínico. En pacientes con insuficiencia renal crónica deberán controlarse regularmente los valores de urea, creatinina, potasio, fósforo y ácido úrico, ya que se han observado discretos aumentos de los valores de estos parámetros tanto en pacientes sometidos a diálisis como en aquellos en prediálisis. Dieta: Al aumentar el hematócrito, se observa una mejoría en el apetito. Por esta razón la ingesta de alimentos en los pacientes en tratamiento con HEMAX® suele aumentar. En estas circunstancias debe tenerse cautela con los valores de potasio, ya que pueden aumentar como consecuencia de la mayor ingesta de alimentos. Manejo de la diálisis: El tratamiento con HEMAX® provoca un aumento del hematócrito con disminución del volumen plasmático que puede afectar la eficacia de la diálisis. Deben realizarse ajustes de la diálisis para impedir el aumento de los valores de urea, fósforo, potasio y creatinina. En algunas ocasiones, puede ser necesario aumentar la dosis de heparina durante la diálisis para prevenir la obstrucción de la fístula.
Advertencias.
Insuficiencia renal: En dos estudios clínicos los pacientes experimentaron mayor riesgo de mortalidad y eventos cardiovasculares serios cuando se les administraron agentes estimulantes de la eritropoyesis, buscando mayores niveles de hemoglobina en comparación a valores menores (13,5 vs 11,3 g/dl; 14 vs 10 g/dl). Se recomienda individualizar la dosis con el objetivo de alcanzar y mantener los niveles de hemoglobina en el rango de 10 a 12 g/dl. Pacientes con diagnóstico de cáncer: El uso de agentes estimulantes de la eritropoyesis acortó la sobrevida general y/o incrementó el riesgo de progresión tumoral o recurrencia en estudios clínicos en pacientes con cáncer de mama, de cabeza y cuello, linfoides, de pulmón (de células no-pequeñas) y cervicales. Para minimizar estos riesgos, al igual que el riesgo de eventos serios cardiovasculares, se recomienda utilizar la dosis más baja para evitar la transfusión sanguínea. Con el objetivo de minimizar los riesgos mencionados el nivel de hemoglobina no debe superar los 12 g/dl. Se recomienda utilizar sólo para el tratamiento de anemia asociada a quimioterapia mielosupresora concomitante y discontinuar su uso luego de haber completado el ciclo de quimioterapia. No se recomienda utilizar eritropoyetinas en pacientes con tratamiento quimioterápico en los cuales se prevea curación. Pacientes que recibieron agentes estimulantes de la eritropoyesis en forma preoperatoria para reducir el número de transfusiones alogénicas: se ha reportado una mayor incidencia de trombosis venosa profunda en pacientes que recibieron agentes estimulantes de la eritropoyesis sin anticoagulación profiláctica. La profilaxis anticoagulante debe ser considerada cuando se indique un agente estimulante de la eritropoyesis para reducir el número de transfusiones alogénicas.
Conservación.
Inyectable Liofilizado: conservar en lugar fresco y seco, a temperatura que no supere los 25°C. Solución inyectable: Conservar en heladera, a temperatura entre 2°C y 8°C. Evitar la luz solar directa durante el almacenamiento. No congelar. Reconstituir con agua estéril para inyectables, usp. Una vez reconstituida la solución, usar inmediatamente.
Sobredosificación.
No se ha definido aún la dosis máxima de HEMAX® que se puede administrar ya sea en bolo o en infusión. Se han utilizado dosis de hasta 1.500 U/Kg tres veces por semana o hasta 60.000 UI/semana en adultos sin encontrarse efectos tóxicos directos. El tratamiento con HEMAX® puede provocar poliglobulia y los pacientes pueden referir síntomas relacionados a la misma, como cefalea, somnolencia, acúfenos, mareos, etc. Ante esta situación se sugiere realizar una flebotomía con el fin de reducir el hematócrito. Ante la eventualidad de una sobredosificación, concurrir al Hospital más cercano o comunicarse con algún Centro de Toxicología. En Argentina: Hospital de Niños Dr. Ricardo Gutiérrez: (011) 4962-6666/2247. Hospital Posadas: (011) 4654-6648/7777.
Presentación.
Inyectable liofilizado: HEMAX® 1000 UI/ml, HEMAX® 10000 UI/ml, HEMAX® 20000 UI/ml, HEMAX® 40000 UI/ml. Envases conteniendo: 1 vial con polvo liofilizado + 1 ampolla con 1 ml de agua estéril para inyectables + 1 jeringa descartable + 2 agujas estériles descartables. HEMAX® 2000 UI/2ml, HEMAX® 3000 UI/2 ml, HEMAX® 4000 UI/2 ml. Envases conteniendo: 1 vial con polvo liofilizado + 1 ampolla con 2 ml de agua estéril para inyectables + 1 jeringa descartable + 2 agujas estériles descartables.
Revisión.
Septiembre 2009.