SUTENT
PFIZER
Agente antineoplásico.
Composición.
Cada cápsula de SUTENT 12,5 mg contiene: SUNITINIB (como sal L-malato) 12,5 mg. Manitol 80,000 mg. Croscarmelosa sódica 6,600 mg. Povidona K-25 5,600 mg. Estearato de magnesio 1,100 mg. Cada cápsula de SUTENT 25 mg contiene: SUNITINIB (como sal L-malato) 25,0 mg. Manitol 39,663 mg. Croscarmelosa sódica 5,010 mg. Povidona K-25 4,175 mg. Estearato de magnesio 1,252 mg. Cada cápsula de SUTENT 50 mg contiene: SUNITINIB (como sal L-malato) 50,0 mg. Manitol 79,326 mg. Croscarmelosa sódica 10,020 mg. Povidona K-25 8,350 mg. Estearato de magnesio 2,504 mg.
Farmacología.
Descripción: SUTENT (L-malato de sunitinib) es suministrado como cápsulas duras recubiertas e impresas conteniendo la cantidad de L-malato de sunitinib equivalente a 12,5 mg, 25 mg o 50 mg de sunitinib base libre, conjuntamente con manitol, croscarmelosa sódica, povidona K-25 y estearato de magnesio como ingredientes inactivos. SUTENT, L-malato de sunitinib, de administración oral, es un inhibidor multi-cinasa que actúa sobre varios receptores de la tirosina cinasa. El L-malato de sunitinib se denomina químicamente como la sal (1:1) del ácido 2(S)-hidroxibutanodioico (ácido L-málico) con la N-[2-(dietilamino) etil]-5-[(Z)-(5-fluoro-1,2-dihidro-2- oxo-3H-indol-3-iliden) metil]-2,4-dimetil-1H-pirrol-3-carboxamida). La fórmula molecular es C22H27FN4O2 • C4H6O5 y el peso molecular es 532,6 Daltons. La estructura química del L-malato de sunitinib es: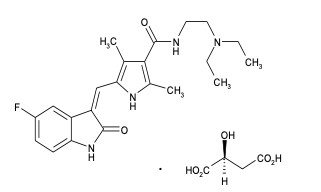
El L-malato de sunitinib es un polvo naranja a amarillo con un pKa de 8,95. La solubilidad del L-malato de sunitinib en medios acuosos por encima del rango de pH 1,2 a pH 6,8 es superior a 25 mg/mL. El logaritmo del coeficiente de distribución (octanol/agua) de pH 7 es 5,2.Propiedades famacodinámicas: Mecanismo de acción: El L-malato de sunitinib es una molécula pequeña que inhibe múltiples receptores de la tirosina cinasa (RTKs), algunos de los cuales están implicados en el crecimiento tumoral, la angiogénesis patológica y la progresión metastásica del cáncer. Se evaluó la actividad inhibitoria de sunitinib contra una variedad de cinasas ( > 80 cinasas) y se lo identificó como un inhibidor de los receptores del factor de crecimiento derivado de plaquetas (PDGFRa y PDGFRb), receptores del factor de crecimiento endotelial vascular (VEGFR1, VEGFR2 y VEGFR3), receptor del factor de células madre (sigla en inglés: SCF) KIT, receptor tirosina cinasa-3 tipo fms (FLT3), receptor de los factores estimulantes de colonias tipo 1 (sigla en inglés: CSF-1R), y el receptor del factor neurotrófico derivado de células gliales (RET). La inhibición por parte de sunitinib de la actividad de estos RTKs se ha demostrado en los ensayos celulares y bioquímicos, y la inhibición de la función se ha demostrado en los ensayos de proliferación celular. El metabolito principal presenta una potencia similar comparada con sunitinib en los ensayos celulares y bioquímicos. Sunitinib inhibió la fosforilación de RTKs múltiples (PDGFRb, VEGFR2, KIT) en xenoinjertos tumorales que expresan objetivos de RTK in vivo y demostró la inhibición del crecimiento tumoral o regresión del tumor y/o inhibió las metástasis en algunos modelos experimentales de cáncer. Se demostró que sunitinib posee la habilidad para inhibir el crecimiento de células tumorales que expresan blancos irregulares de los RTKs (PDGFR, RET, o KIT) in vitro y para inhibir la angiogénesis tumoral in vivo dependientes de PDGFRb y VEGFR2. Propiedades farmacocinéticas: La farmacocinética del sunitinib y del L-malato de sunitinib se ha evaluado en 135 voluntarios sanos y en 266 pacientes con tumores sólidos. Absorción: Las concentraciones plasmáticas máximas (Cmax) de sunitinib se observan generalmente entre 6 y 12 horas (tiempo máximo de concentración en plasma, Tmax) luego de la administración oral. Las comidas no producen efectos sobre la biodisponibilidad de sunitinib. Sunitinib puede ingerirse con o sin alimentos. Distribución: La unión de sunitinib y su metabolito principal a las proteínas plasmáticas humanas in vitro fue del 95 y 90%, respectivamente, sin dependencia de la concentración en el rango de 100 - 4000 ng/mL. El volumen aparente de distribución (Vd/F) de sunitinib fue de 2230 L. En el rango de dosis de 25 - 100 mg, el ABC versus tiempo y la concentración plasmática máxima (Cmax) aumentan proporcionalmente con la dosis. Metabolismo: Sunitinib se metaboliza principalmente a través de la enzima CYP3A4 del citocromo P450, para producir su metabolito activo principal, el cual es posteriormente metabolizado por la enzima CYP3A4. El metabolito activo principal constituye el 23% al 37% de la exposición total. Eliminación: La principal vía de eliminación es a través de las heces. En un estudio de equilibrio de masa realizado con sunitinib [14C] en humanos, el 61% de la dosis fue eliminada en las heces, la eliminación renal fue del 16% de la dosis administrada. Sunitinib y su metabolito activo principal fueron los principales componentes relacionados con el fármaco identificados en el plasma, orina y heces, representando el 91,5%; 86,4% y 73,8% de la radioactividad en muestras agrupadas, respectivamente. Se identificaron metabolitos menores en la orina y en las heces pero en general no se encontraron en el plasma. El clearance oral total (CL/F) osciló entre 34 y 62 L/h con una variabilidad interpaciente del 40%. Luego de la administración de una dosis oral única a voluntarios sanos, las vidas medias terminales de sunitinib y su metabolito activo principal son de aproximadamente 40 a 60 horas y 80 a 110 horas, respectivamente. Con la administración diaria reiterada, sunitinib se acumula de tres a cuatro veces, mientras que el metabolito principal se acumula de siete a diez veces. Las concentraciones de sunitinib en el estado estable y de su metabolito activo principal se alcanzan dentro de los 10 a 14 días. Para el día 14, las concentraciones plasmáticas combinadas de sunitinib y de su metabolito activo oscilaron entre 62,9 y 101 ng/mL. Ningún cambio significativo en la farmacocinética de sunitinib o del metabolito activo principal se observó con la administración diaria reiterada o con los ciclos reiterados en los regímenes de dosis evaluados. La farmacocinética fue similar en los voluntarios sanos y en las poblaciones de pacientes con tumores sólidos evaluados, incluyendo pacientes con tumores del estroma gastrointestinal (GIST) y carcinoma de células renales. Farmacocinética en poblaciones especiales: Los análisis farmacocinéticos de la población, de acuerdo a los datos demográficos, indican que la edad, el peso corporal, el clearance de creatinina, la raza, el sexo o el puntaje del ECOG (Eastern Cooperative Oncology Group, Grupo Cooperativo de Oncología del Este) no producen efectos clínicamente relevantes sobre la farmacocinética de SUTENT o de su metabolito activo principal. Insuficiencia hepática: Las exposiciones sistémicas después de una única dosis de sunitinib fueron similares en pacientes con deterioro de la función hepática exocrina leve (Clase A de Child-Pugh) o moderado (Clase B de Child-Pugh), comparadas con las de pacientes con función hepática normal. Insuficiencia renal: La exposición sistémica al sunitinib luego de una dosis única de SUTENT fue similar en pacientes con deterioro severo de la función renal (CLcr < 30 ml/min) al observado en aquellos con función renal normal (CLcr > 80 ml/min). Aunque el sunitinib no fue eliminado por hemodiálisis, la exposición sistémica al sunitinib fue 47% menor en pacientes con ERET en hemodiálisis, comparados con pacientes con función renal normal. Niños: No se ha evaluado la farmacocinética de sunitinib en pacientes pediátricos. Ensayos clínicos: Tumores del estroma gastrointestinal: Estudio 1: El estudio 1 (NCT#00075218) fue un estudio internacional de SUTENT de 2 ramas de tratamiento, aleatorizado, doble ciego, controlado con placebo, en pacientes con GIST que habían presentado progresión de la enfermedad durante el tratamiento previo con mesilato de imatinib (imatinib) o que eran intolerantes al imatinib. El objetivo fue comparar el tiempo a la progresión del tumor (sigla en inglés: TTP) en pacientes que estaban recibiendo SUTENT más las mejores medidas de apoyo, versus pacientes que estaban recibiendo placebo, más las mejores medidas de apoyo. Otros objetivos incluyeron sobrevida libre de progresión (sigla en inglés: PFS), tasa de respuesta objetiva (sigla en inglés: ORR), y la sobrevida global (sigla en inglés: OS). Los pacientes fueron aleatorizados (2:1) para recibir 50 mg de SUTENT o placebo, vía oral, una vez al día, en un esquema 4/2 hasta la progresión de la enfermedad o retiro del estudio por otra razón. El tratamiento no fue ciego al momento de la progresión de la enfermedad. A los pacientes aleatorizados inicialmente a la rama placebo se les ofreció luego ser cruzados a un tratamiento abierto con SUTENT, y a los pacientes aleatorizados a SUTENT se les permitió continuar con el tratamiento, de acuerdo a la opinión del investigador. Al momento del análisis intermedio pre-especificado, la población con intención de tratar (sigla en inglés: ITT) incluyó 312 pacientes. Doscientos siete (207) pacientes fueron aleatorizados a la rama SUTENT, y 105 pacientes fueron aleatorizados al grupo placebo. Los datos demográficos de los grupos tratados con SUTENT y con placebo fueron comparables con respecto a la edad (69% versus. 72% < 65 años para SUTENT versus placebo, respectivamente), el sexo (masculino: 64% versus 61%), la raza (blanca, 88% en ambas ramas; asiática, 5% en ambas ramas; negra, 4% en ambas ramas; y el resto no informadas) y el estado general (Escala ECOG 0: 44% versus 46%, ECOG 1: 55% versus 52% y ECOG 2: 1% versus 2%). Los tratamientos previos incluyeron cirugía (94% versus 93%) y radioterapia (8% versus 15%). Los resultados del tratamiento previo con imatinib también fueron comparables entre las ramas e incluyeron intolerancia (4% versus 4%), progresión dentro de los 6 meses de iniciado el tratamiento (17% versus 16%) o progresión después de los 6 meses (78% versus 80%). Se realizó un análisis provisorio y planificado de la seguridad y eficacia luego de que se produjeron 149 eventos de TTP. Se registró una ventaja estadísticamente significativa de SUTENT sobre el placebo en cuanto al TTP, alcanzando el objetivo primario del estudio. Los resultados de eficacia se resumen en la tabla 1 y la curva de Kaplan-Meier para la TTP se incluye en la figura 1.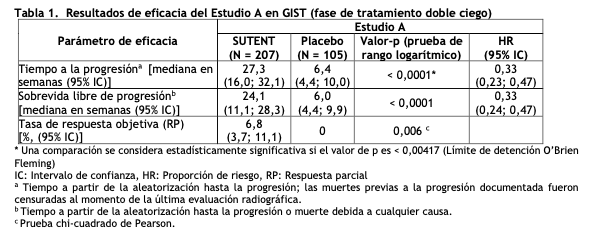
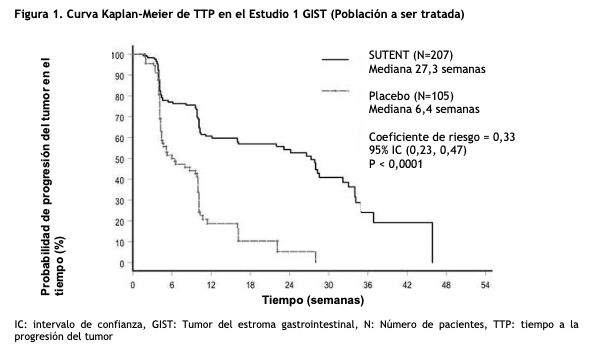
La población ITT final enrolada en la fase de tratamiento doble ciego del estudio incluyó a 243 pacientes aleatorizados al grupo de SUTENT y 118 pacientes aleatorizados al grupo de placebo. Luego de que se cumpliera el objetivo primario del estudio en el análisis intermedio, se divulgó el ciego del estudio, y a los pacientes que estaban en el grupo de placebo se les ofreció recibir tratamiento abierto con SUTENT. De los pacientes inicialmente aleatorizados al placebo, 99 pasaron a la fase de tratamiento abierto con SUTENT. En el análisis final de la OS especificado del protocolo, la mediana de la OS fue de 72,7 semanas en el grupo de SUTENT y de 64,9 semanas en el grupo de placebo [HR= 0,876; IC del 95% (0,679; 1,129)]. Estudio 2: El estudio 2 fue un estudio con aumento de dosis, abierto, multicéntrico, de una sola rama de tratamiento, realizado en pacientes con GIST luego de la progresión o intolerancia al imatinib. Luego de la identificación del régimen recomendado (50 mg una vez al día en un esquema 4/2), 55 pacientes en este estudio recibieron una dosis de 50 mg de SUTENT en un esquema 4/2 de tratamiento. Se observaron respuestas parciales en 5 de 55 pacientes [9,1% porcentaje de RP, 95% IC (3,0; 20,0)]. Carcinoma de células renales: Carcinoma de células renales, en pacientes vírgenes de tratamiento (naïve): El Estudio 3 (NCT#00083889) fue un estudio multicéntrico internacional y aleatorizado de comparación de SUTENT como agente único con IFN-a en pacientes con carcinoma de células renales, vírgenes de tratamiento (naïve). Su objetivo fue comparar la sobrevida libre de progresión (PFS) en pacientes que recibían SUTENT versus pacientes que recibían IFN-a. Otros puntos finales incluyeron la tasa de respuesta objetiva (ORR), la sobrevida global (OS) y el perfil de seguridad. Setecientos cincuenta (750) pacientes fueron aleatorizados (1:1) para recibir o bien 50 mg de SUTENT una vez por día según el esquema 4/2 o bien IFN-a administrado por vía subcutánea a 9 millones de unidades internacionales (MUI), 3 veces por semana. Los pacientes fueron tratados hasta que la enfermedad progresó o se retiraron del estudio. La población con intención de tratar (ITT) incluyó 750 pacientes; 375 fueron aleatorizados a SUTENT y 375 fueron aleatorizados a IFN-a. Los datos demográficos de los grupos tratados con SUTENT e IFN-a fueron comparables con respecto a la edad (59% versus 67% < 65 años para SUTENT versus IFN-a, respectivamente), el sexo (masculino: 71% versus 72%), la raza (blanca, 94% versus 91%; asiática, 2% versus 3%; negra 1% versus 2%; y el resto no informada) y el estado general (escala ECOG 0: 62% versus 61%; ECOG 1, 38% en cada rama y ECOG 2, 0 versus 1%). Los tratamientos anteriores incluyeron nefrectomía (91% versus 89%) y radioterapia (14% en cada rama). La enfermedad metastásica más común presente en la etapa de selección fue la de pulmón (78% versus 80%, respectivamente) seguida por la de ganglios linfáticos (58% versus 53%, respectivamente) y la ósea (30% en cada rama). La mayoría de los pacientes presentaba múltiples (2 o más) sitios metastásicos en la visita basal (80% versus 77%, respectivamente). Hubo una ventaja estadísticamente significativa de SUTENT sobre IFN-a en el objetivo PFS (ver tabla 2 y figura 2). En los factores de estratificación especificados previamente para lactato dehidrogenasa (LDH) ( > 1,5 el LSN versus ≤1,5 el LSN), estado general según la escala ECOG (0 versus 1) y nefrectomía previa (sí versus no), la proporción de riesgo (hazard ratio) favoreció a SUTENT sobre IFN-a. La ORR fue superior en la rama de SUTENT (ver tabla 2).
En el análisis final de la OS especificado en el protocolo, la mediana de la OS fue de 114,6 semanas en el grupo de SUTENT y de 94,9 semanas en el grupo de IFN-a [HR= 0,821; IC del 95% (0,673; 1,001)]. La mediana de la OS en el grupo de IFN-a incluye a 25 pacientes que discontinuaron el tratamiento con IFN-a debido a progresión de la enfermedad y que pasaron al tratamiento con SUTENT, y a 121 pacientes (32%) del grupo de IFN-a que recibieron tratamiento para el cáncer con SUTENT posterior al estudio. Carcinoma de células renales refractario a las citoquinas: El uso de SUTENT como agente único en el tratamiento del carcinoma de células renales refractario a las citoquinas se investigó en 2 estudios multicéntricos de rama única. Todos los pacientes que participaron en estos estudios fracasaron con los tratamientos previos basados en citoquinas. En el Estudio 4 (NCT#00077974), el fracaso a las terapias previas con citoquinas se basó en pruebas radiográficas de progresión de la enfermedad definida por criterios RECIST [Response Evaluation Criteria In Solid Tumors (criterios de evaluación de respuesta en tumores sólidos)] o los criterios de la Organización Mundial de la Salud (OMS) durante o dentro de los 9 meses de completar un tratamiento con citoquinas (IFN-a, interleuquina 2, o IFN-a más interleuquina 2; los pacientes que fueron tratados con IFN-a solo, debían haber recibido tratamiento por al menos 28 días). En el Estudio 5 (NCT#00054886), el fracaso a la terapia previa con citoquinas se definió como progresión de la enfermedad o toxicidad no aceptable relacionada con el tratamiento. El punto final para ambos estudios fue la ORR (Tasa de Respuesta Objetiva). La duración de la respuesta (DR) también fue evaluada. Ciento seis (106) pacientes participaron del Estudio 4 y 63 pacientes participaron del Estudio 5. Los pacientes recibieron 50 mg de SUTENT en un esquema 4/2. La terapia se continuó hasta que los pacientes reunieran los criterios para la suspensión o tuvieran enfermedad progresiva. Las condiciones de los resultados del ECOG [Grupo Cooperativo de Oncología del Este (Eastern Cooperative Oncology Group)], raza, sexo y edad inicial de los pacientes fueron comparables entre los Estudios 4 y 5. Aproximadamente el 86% - 94% de los pacientes en los dos estudios eran de raza blanca. Los hombres componían el 65% de la población agrupada. La edad media fue de 57 años y osciló entre los 24 y 87 años en los estudios. Todos los pacientes tuvieron condiciones de resultados ECOG < 2 en la visita de selección. La malignidad basal y los antecedentes de tratamientos previos del paciente fueron comparables entre los Estudios 4 y 5. A través de los dos estudios, el 95% de la población agrupada de pacientes tuvieron al menos algunos componentes histológicos de células claras. En el Estudio 4 se requirió que todos los pacientes tuviesen un componente histológico de células claras. La mayoría de los pacientes que participaron de estos estudios (97% de la población agrupada) habían sido sometidos a una nefrectomía; se requirió nefrectomía previa en los pacientes que participaron del Estudio 4. Todos los pacientes habían recibido un régimen previo con citoquinas. La enfermedad metastásica presente al momento del ingreso al estudio incluyó metástasis en el pulmón en el 81% de los pacientes. Las metástasis en el hígado fueron más comunes en el Estudio 4 (27% versus 16% en el Estudio 5) y las metástasis óseas fueron más comunes en el Estudio 5 (51% versus 25% en el Estudio 4); el 52% de los pacientes en la población agrupada presentó por lo menos 3 sitios metastásicos. Los pacientes con metástasis cerebral o enfermedad leptomeníngea conocidas fueron excluidos de ambos estudios. Los datos de la ORR y DR de los Estudios 4 y 5 se proporcionan en la tabla 3.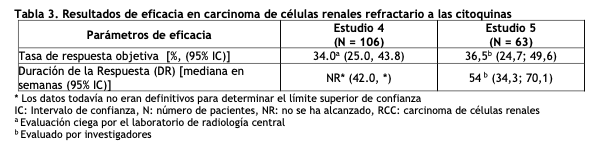
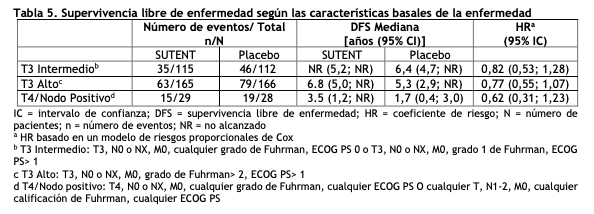
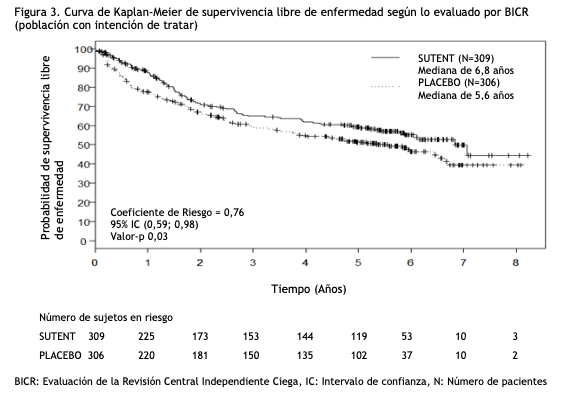
Se informaron 36 respuestas parciales (RPs) en el Estudio 4, de acuerdo a la evaluación del laboratorio de radiología central, con una ORR del 34,0% (95% IC 25,0%, 43,8%). Se observaron 23 RPs en el Estudio 5, de acuerdo a la evaluación de los investigadores, con una ORR del 36,5% (95% IC 24,7% - 49,6%). La mayoría ( > 90%) de las respuestas de enfermedad objetivas fueron observadas durante los primeros cuatro ciclos; la última respuesta informada se observó en el ciclo 10. La información sobre la duración de la respuesta (DR) del Estudio 4 es prematura dado que sólo 9 de los 36 pacientes (25%) que respondieron al tratamiento habían experimentado progresión de la enfermedad o fallecido al momento del corte de datos. Tratamiento adyuvante para Carcinoma de células renales (RCC): En el tratamiento adyuvante, SUTENT se investigó en S-TRAC (NCT # 00375674), un ensayo multicéntrico, internacional, aleatorizado, doble ciego, controlado con placebo en pacientes con alto riesgo de RCC recurrente después de la nefrectomía. Se requirió que los pacientes tuvieran una histología de células claras y un alto riesgo de recurrencia definido como tumores ≥T3 y/o N +. Seiscientos quince (615) pacientes fueron aleatorizados 1:1 para recibir 50 mg de SUTENT una vez al día en un esquema 4/2 o placebo. Los pacientes fueron tratados durante 9 ciclos (aproximadamente 1 año), o hasta la recurrencia de la enfermedad, toxicidad inaceptable o el retiro del consentimiento. Los datos demográficos fueron generalmente comparables entre los grupos SUTENT y placebo con respecto a la edad (mediana de edad 58 años), género (73% hombres) y raza (84% caucásicos, 12% asiáticos y 4% otros). En la aleatorización, la mayoría de los pacientes tenían un estado funcional ECOG de 0 (74% SUTENT y 72% placebo). El resto de los pacientes tenía un estado de rendimiento ECOG de 1; 1 paciente en SUTENT tuvo un estado de rendimiento de 2. El mayor criterio de resultado de eficacia fue la supervivencia libre de enfermedad (DFS por sus siglas en inglés) en pacientes que recibieron SUTENT versus placebo según la evaluación de la revisión central independiente ciega (BICR). La supervivencia global fue un punto final adicional. Hubo una mejoría estadísticamente significativa en la DFS en pacientes que fueron tratados con SUTENT en comparación con placebo (Tabla 4 y Figura 3). Los análisis de subgrupos preespecificados se presentan en la Tabla 5. En el momento del análisis de DFS, los datos de supervivencia global no estaban completos, con 141/615 (23%) muertes de pacientes.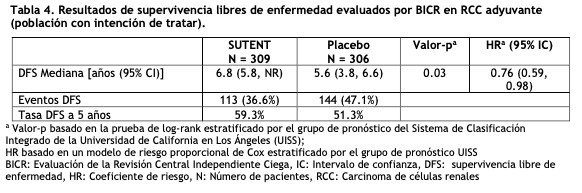
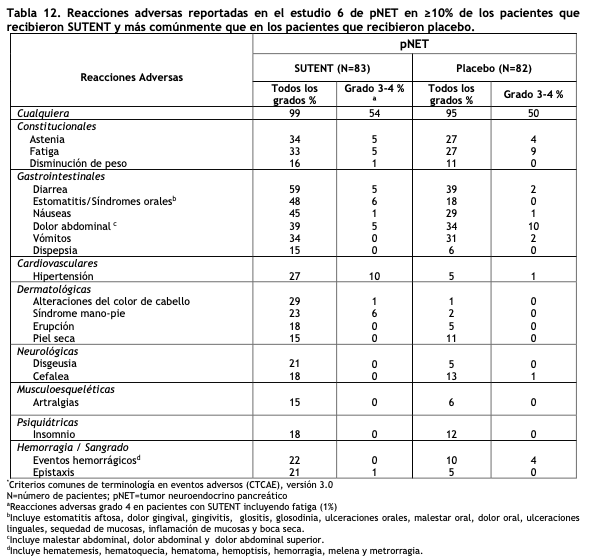
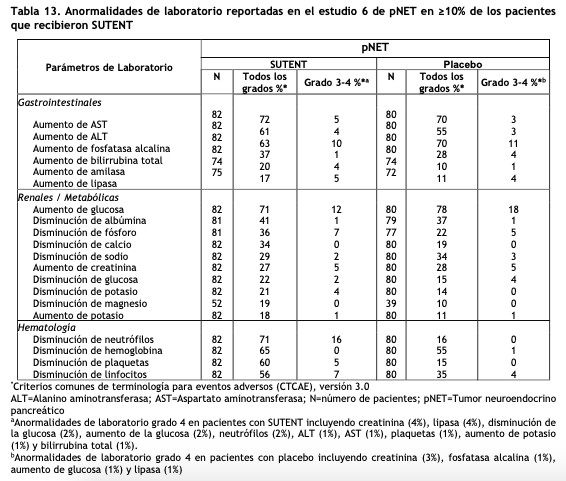
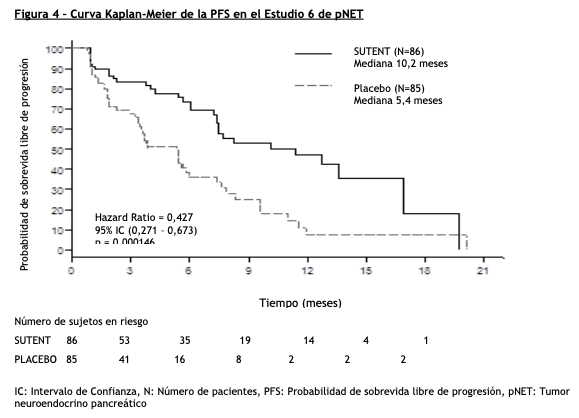
Tumores neuroendocrinos pancreáticos (pNET): El Estudio 6 (NCT#00428597) fue un estudio multicéntrico, internacional, aleatorizado, doble ciego y controlado con placebo de sunitinib como agente único en pacientes con pNET no resecable. Se requirió que los pacientes tuvieran documentada progresión, según RECIST, durante los 12 meses previos y se les distribuyó en forma aleatoria (1:1) para recibir sunitinib 37,5 mg una vez al día sin periodo de descanso programado (N=86) o placebo (N=85). El objetivo primario fue comparar la sobrevida libre de progresión (PFS) en pacientes que recibieron sunitinib versus pacientes que recibieron placebo. Otros objetivos del estudio fueron la sobrevida global (OS), la tasa de respuesta objetiva (ORR) y la seguridad. En este estudio se permitió el uso de análogos de somatostatina. Los datos demográficos fueron comparables entre los grupos de sunitinib y placebo. Adicionalmente, el 49% de los pacientes con sunitinib tuvieron tumores no funcionales versus el 52% de los pacientes con placebo y el 92% de los pacientes en ambos grupos tuvieron metástasis hepáticas. Un total del 66% de los pacientes con sunitinib habían recibido tratamiento sistémico previo en comparación con el 72% de los pacientes con placebo. Además, el 35% de los pacientes con sunitinib habían recibido análogos de somatostatina en comparación con el 38% de los pacientes con placebo. Los pacientes fueron tratados hasta la progresión de la enfermedad o retiro del estudio. Ante la progresión de la enfermedad o el cierre del estudio, se ofreció a los pacientes el acceso a SUTENT en un estudio de extensión separado. Según la recomendación del Comité Independiente de Monitoreo de Datos, el estudio fue terminado prematuramente antes del análisis interino pre-especificado. Esto puede haber llevado a una sobreestimación de la magnitud del efecto sobre la PFS. Se observó una mejoría clínicamente significativa en la PFS para sunitinib sobre placebo evaluada tanto por el investigador como por una evaluación independiente. Se observó un hazard ratio a favor de SUTENT en todos los subgrupos de las características basales evaluadas. Los datos de OS no eran definitivos en el momento del análisis. Hubo 9 muertes en el grupo de sunitinib y 21 muertes en el grupo placebo. Se observó una diferencia estadísticamente significativa en la ORR a favor de sunitinib frente al placebo. Los resultados de eficacia pueden verse en la tabla 6 y en la figura 4.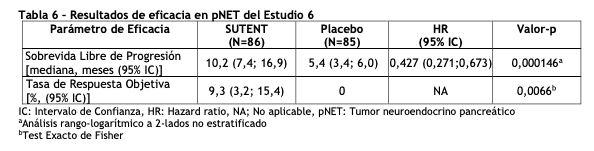
Toxicologia No Clinica: Carcinogénesis, mutagénesis, deterioro de la fertilidad: Se ha evaluado el potencial carcinogénico de sunitinib en dos especies: ratones transgénicos rasH2 y ratas Sprague-Dawley. Hubo hallazgos positivos similares en ambas especies. En los ratones transgénicos rasH2, se observaron carcinomas gastroduodenales y/o hiperplasia de la mucosa gástrica, al igual que un aumento en la incidencia de hemangiosarcomas de fondo a dosis de ≥25 mg/kg/día luego de la administración de una dosis diaria de sunitinib en estudios de uno o seis meses de duración. No se observaron cambios proliferativos en ratones transgénicos rasH2 a 8 mg/kg/día. De manera similar, en un estudio de 2 años de carcinogenicidad en ratas, la administración de sunitinib en ciclos de 28 días seguidos por períodos de 7 días sin dosis dio como resultado hallazgos de carcinoma duodenal a dosis más bajas como 1 mg/kg/día (aproximadamente 0,9 veces el ABC en pacientes a los que se administra DDR de 50 mg/día). A la dosis alta de 3 mg/kg/día (aproximadamente 7,8 veces el ABC en pacientes al DDR de 50 mg/día) la incidencia de tumores duodenales se vio aumentada y estuvo acompañada de hallazgos de hiperplasia de las células de la mucosa gástrica y un aumento en la incidencia de feocromocitoma e hiperplasia de la glándula suprarrenal. Sunitinib no causó daños genéticos cuando fue testeado en ensayos in vitro [mutación bacteriana (Test de AMES), aberración cromosómica de linfocitos humanos] y en pruebas in vivo de micronúcleos de médula ósea de rata. En un estudio de fertilidad femenina y desarrollo embrionario temprano, a las ratas hembras se les administró sunitinib oral (0,5; 1,5; 5 mg/kg/día) durante 21 días antes del apareamiento y durante 7 días después del apareamiento. Se observó pérdida de preimplantación en las hembras a las que se les administró 5 mg/kg/día (aproximadamente 5 veces el ABC en pacientes a los que se administró el DDR de 50 mg/día). No se observaron efectos adversos sobre la fertilidad a dosis ≤1.5 mg/kg/día (aproximadamente 1 vez el ABC para DDR de 50 mg/día). Además, los efectos en el sistema reproductor femenino fueron identificados en un estudio en monos de dosis orales repetidas de 3 meses de duración (2, 6, 12 mg/kg/día). Los cambios ováricos (disminución del desarrollo folicular) fueron observados con 12 mg/kg/día (aproximadamente 5,1 veces el ABC en los pacientes a los que se les administró la DDR), mientras que los cambios uterinos (atrofia endometrial) se notaron con ≥ 2 mg/kg/día (aproximadamente 0,4 veces el ABC en pacientes a los que se les administró la DDR). Con el agregado de atrofia vaginal, los efectos ováricos y uterinos se reprodujeron con 6 mg/kg/día en un estudio en monos de 9 meses de duración (0,3; 1,5 y 6 mg/kg/día administrados durante 28 días seguidos con un descanso de 14 días). En un estudio de fertilidad masculina, no se observaron efectos en la reproducción de ratas machos con dosis de (1, 3 o 10 mg/kg/día) durante 58 días previos al apareamiento con hembras no tratadas. La fertilidad, la copulación, los índices de concepción y la evaluación de los espermas (morfología, concentración y motilidad) no se vieron afectadas por sunitinib con dosis ≤ 10 mg/kg/día (aproximadamente 26 veces la ABC en pacientes a los que se les administró la DDR).
Indicaciones.
SUTENT está indicado para el tratamiento de los tumores del estroma gastrointestinal (sigla en inglés: GIST) después de la progresión de la enfermedad durante el tratamiento con mesilato de imatinib o de la intolerancia al mismo. SUTENT está indicado para el tratamiento del carcinoma de células renales avanzado (sigla en inglés: RCC). SUTENT está indicado para el tratamiento adyuvante de pacientes adultos con alto riesgo de carcinoma de células renales recurrente después de la nefrectomía. SUTENT está indicado para el tratamiento de tumores neuroendocrinos pancreáticos (pNET bien diferenciados, no resecables, localmente avanzados o metastásicos, con progresión de la enfermedad).
Dosificación.
La dosis recomendada de SUTENT para los tumores del estroma gastrointestinal (GIST) y el carcinoma de células renales avanzado (RCC) es una dosis oral de 50 mg administrada una vez al día, en un esquema de 4 semanas de tratamiento seguido por 2 semanas de descanso (esquema 4/2) para constituir un ciclo completo de 6 semanas. La dosis recomendada de SUTENT para el tratamiento adyuvante del carcinoma de células renales (RCC) es una dosis oral de 50 mg administrada una vez al día, en un esquema de 4 semanas de tratamiento seguido por 2 semanas de descanso (esquema 4/2) para constituir un ciclo completo de 6 semanas. La dosis recomendada de SUTENT para pNET es de 37,5 mg por vía oral una vez al día, sin período de descanso programado. SUTENT puede ser ingerido con o sin alimentos. Si se pierde una dosis, el paciente no debe recibir una dosis adicional. El paciente debe recibir la dosis usual prescripta el día siguiente. Puede ser necesario interrumpir la administración según la seguridad y la tolerabilidad individual. Modificación de la dosis: Se recomienda la interrupción de la dosis y/o modificaciones de la dosis, incrementos o reducciones, a razón de 12,5 mg en base a la seguridad y tolerancia individual. En el caso de pNET, se pueden aplicar modificaciones de la dosis con variaciones de 12,5 mg en base a la seguridad y la tolerabilidad individual. La dosis máxima administrada en el estudio de pNET fue de 50 mg al día. La dosis mínima administrada en el estudio para tratamiento adyuvante de carcinoma de células renales (RCC) fue de 37,5 mg. Los inhibidores potentes de la CYP3A4 tales como el ketoconazol pueden aumentar las concentraciones plasmáticas de sunitinib. Se recomienda la selección de una medicación concomitante alternativa con potencial de inhibición enzimática nulo o mínimo. Se deberá considerar una reducción de la dosis de SUTENT a un mínimo de 37,5 mg diarios en GIST y RCC o 25 mg diarios en pNET, si SUTENT debe ser co-administrado con un inhibidor potente de la CYP3A4 (ver Características farmacológicas y Advertencias y Precauciones especiales de uso, Interacción con otros medicamentos y otras formas de interacción). Los inductores de la CYP3A4 tales como rifampicina pueden disminuir las concentraciones plasmáticas de sunitinib. Se recomienda la selección de una medicación concomitante alternativa con potencial de inducción enzimática nulo o mínimo. Se deberá considerar un incremento de la dosis de SUTENT a un máximo de 87,5 mg diarios en GIST y RCC o 62,5 mg diarios en pNET, si SUTENT debe ser co-administrado con un inductor de la CYP3A4. Si la dosis se incrementa, deberá monitorearse al paciente cuidadosamente a fin de evaluar la presencia de toxicidad (ver Características farmacológicas y Advertencias y Precauciones especiales de uso, Interacción con otros medicamentos y otras formas de interacción).
Contraindicaciones.
El uso de SUTENT está contraindicado en pacientes con hipersensibilidad al L-malato de sunitinib o a algún otro componente de SUTENT.
Reacciones adversas.
Las siguientes reacciones adversas se detallan en otras secciones de ese prospecto. Hepatotoxicidad (ver Advertencias y Precauciones). Eventos cardiovasculares (ver Advertencias y Precauciones). Prolongación del intervalo QT y torsión de puntas (ver Advertencias y Precauciones). Hipertensión (ver Advertencias y Precauciones). Eventos hemorrágicos (ver Advertencias y Precauciones). Síndrome de Lisis Tumoral (SLT) (ver Advertencias y Precauciones). Microangiopatía trombótica (ver Advertencias y Precauciones). Proteinuria (ver Advertencias y Precauciones). Toxicidades dermatológicas (ver Advertencias y Precauciones). Disfunción tiroidea (ver Advertencias y Precauciones). Hipoglucemia (ver Advertencias y Precauciones). Osteonecrosis de la mandíbula (ONM) (ver Advertencias y Precauciones). Cicatrización de heridas (ver Advertencias y Precauciones). Experiencia en Estudio Clínicos: Debido a que los ensayos clínicos se realizan en condiciones que difieren ampliamente; por lo tanto, las tasas de reacciones adversas observadas en los ensayos clínicos de una droga no pueden compararse directamente con las tasas obtenidas en los ensayos clínicos de otras drogas, y quizás no reflejen las tasas observadas en la práctica. Los datos descriptos en la sección Advertencias y Precauciones, ilustran la exposición a SUTENT (N=7527) en GIST, RCC avanzado, tratamiento adyuvante para RCC, y pNET (ver Advertencias y Precauciones). En esta base de datos, Las reacciones adversas más frecuentes (≥25%) fueron fatiga, astenia, diarrea, mucositis/estomatitis, náuseas, disminución del apetito/anorexia, vómitos, dolor abdominal, síndrome mano-pie, hipertensión, sangrado, alteraciones del gusto/disgeusia, dispepsia, y trombocitopenia. Los datos a continuación reflejan la exposición a SUTENT en 966 pacientes que participaron en la fase de tratamiento doble ciego de un ensayo controlado con placebo (n=202) para el tratamiento de GIST, RCC avanzado (n=375), tratamiento adyuvante para RCC (n=306) y pNET (n=83) (ver Ensayos clinicos). Tumor del estroma gastrointestinal: La seguridad de SUTENT se evaluó en el estudio 1 aleatorizado, controlado con placebo a doble ciego, en el cual los pacientes previamente tratados para GIST, recibieron 50 mg por día de SUTENT, en un esquema 4/2 (n=202) o placebo (n=102). La duración media del tratamiento en el estudio ciego fue de 2 ciclos para los pacientes con SUTENT (promedio 3,0 en un rango de 1-9) y un ciclo (promedio 1,8 en un rango de 1-6) para los pacientes con placebo, en el momento del análisis intermedio. Se realizaron reducciones de dosis en 23 pacientes (11%) con SUTENT y en ninguno con placebo. Interrupciones de las dosis ocurrieron en 59 pacientes (29%) con SUTENT y 31 pacientes (30%) con placebo. Las tasas de reacciones adversas no fatales emergentes del tratamiento que ocasionaron una discontinuación permanente fueron del 7% y 6% en los grupos de SUTENT y placebo, respectivamente. La mayoría de las reacciones adversas emergentes del tratamiento en ambos grupos de estudio fueron de grado 1 o 2 de gravedad. Las reacciones adversas emergentes del tratamiento de grado 3 o 4 se informaron en el 56% versus 51% de los pacientes con SUTENT versus placebo, respectivamente, en la fase de tratamiento doble ciego del estudio. La tabla 7 compara la incidencia de las reacciones adversas comunes emergentes del tratamiento (≥10%) en los pacientes que recibieron SUTENT y que fueron informadas más comúnmente que en pacientes que recibieron placebo.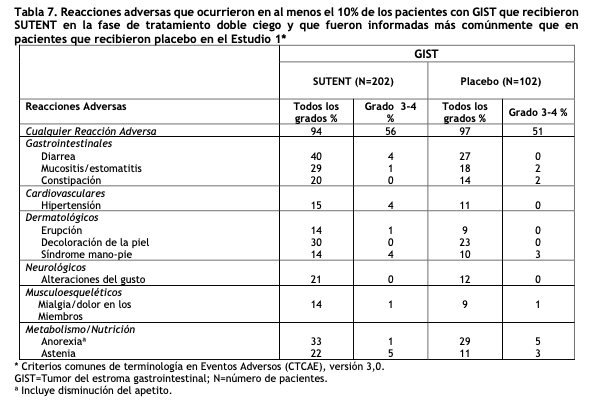
En la fase de tratamiento doble ciego del Estudio 1 de GIST, se informó dolor oral diferente de la mucositis/estomatitis en 12 pacientes (6%) con SUTENT versus 3 (3%) con placebo. Se observaron cambios de color del cabello en 15 pacientes (7%) con SUTENT versus 4 (4%) con placebo. Se informó alopecía en 10 pacientes (5%) con SUTENT versus 2 (2%) con placebo. La tabla 8 proporciona las anormalidades de laboratorio comunes, emergentes del tratamiento (≥ 10%).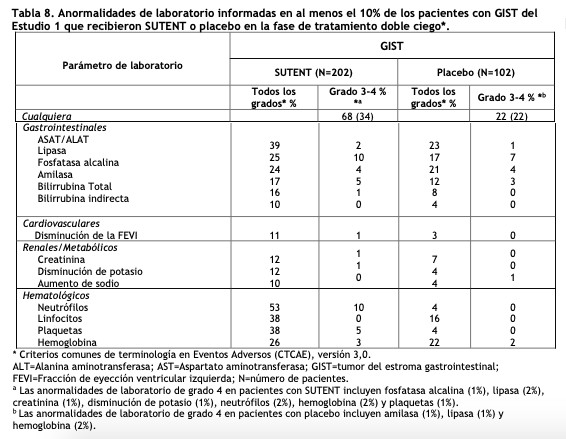
Luego de un análisis intermedio, se divulgó el ciego del estudio, y a los pacientes que estaban en el grupo de placebo se les ofreció la oportunidad de recibir tratamiento abierto con SUTENT (ver Ensayos clínicos). En los 241 pacientes aleatorizados al grupo de SUTENT, incluidos 139 que recibieron SUTENT en las fases de tratamiento doble ciego y abierta, la mediana de la duración del tratamiento con SUTENT fue de 6 ciclos (media 8,5; rango 1-44). En los 255 pacientes que en última instancia recibieron el tratamiento abierto con SUTENT, la mediana de la duración del tratamiento del estudio fue de 6 ciclos (media 7,8; rango 1 - 37) desde el momento de la divulgación del ciego. En total, 118 pacientes (46%) debieron interrumpir el tratamiento y 72 pacientes (28%) requirieron una reducción de la dosis. La incidencia de reacciones adversas emergentes del tratamiento que causó la discontinuación permanente fue del 20%. Los eventos adversos de grado 3 o 4 relacionados con el tratamiento que fueron frecuentes en los pacientes tratados con SUTENT en la fase de tratamiento abierto fueron fatiga (10%), hipertensión (8%), astenia (5%), diarrea (5%), síndrome de mano-pie (5%), náuseas (4%), dolor abdominal (3%), anorexia (3%), mucositis (2%), vómitos (2%) e hipotiroidismo (2%). Carcinoma de células renales avanzado: La seguridad de SUTENT se evaluó en el Estudio 3; doble ciego y activo controlado; en el cual los pacientes con RCC metastásico o avanzado, sin tratamiento previo, recibieron 50 mg por día de SUTENT en un esquema 4/2 (n=375) o 9 millones de unidades internacionales (UI) de IFN-a (n=360) La mediana de la duración del tratamiento fue de 11,1 meses (rango: 0,4 - 46,1) con SUTENT y de 4,1 meses (rango: 0,1 - 45,6) con IFN-a. Se produjeron interrupciones de la dosis en 202 pacientes (54%) tratados con SUTENT y en 141 pacientes (39%) tratados con IFN-a. Se produjeron reducciones de la dosis en 194 pacientes (52%) tratados con SUTENT y en 98 pacientes (27%) tratados con IFN-a. Los índices de discontinuación a causa de reacciones adversas fueron 20% para SUTENT y 24% para el IFN-a.La mayoría de las reacciones adversas emergentes del tratamiento en ambos brazos del estudio fueron de severidad de grado 1 o 2. Las reacciones adversas emergentes del tratamiento de grado 3 o 4 fueron reportadas en 77% vs 55% de los pacientes tratados con SUTENT versus pacientes tratados con IFN-a, respectivamente. En la tabla 9 se compara la incidencia de las reacciones adversas frecuentes (≥10%) emergentes del tratamiento en pacientes tratados con SUTENT versus pacientes tratados con IFN-a.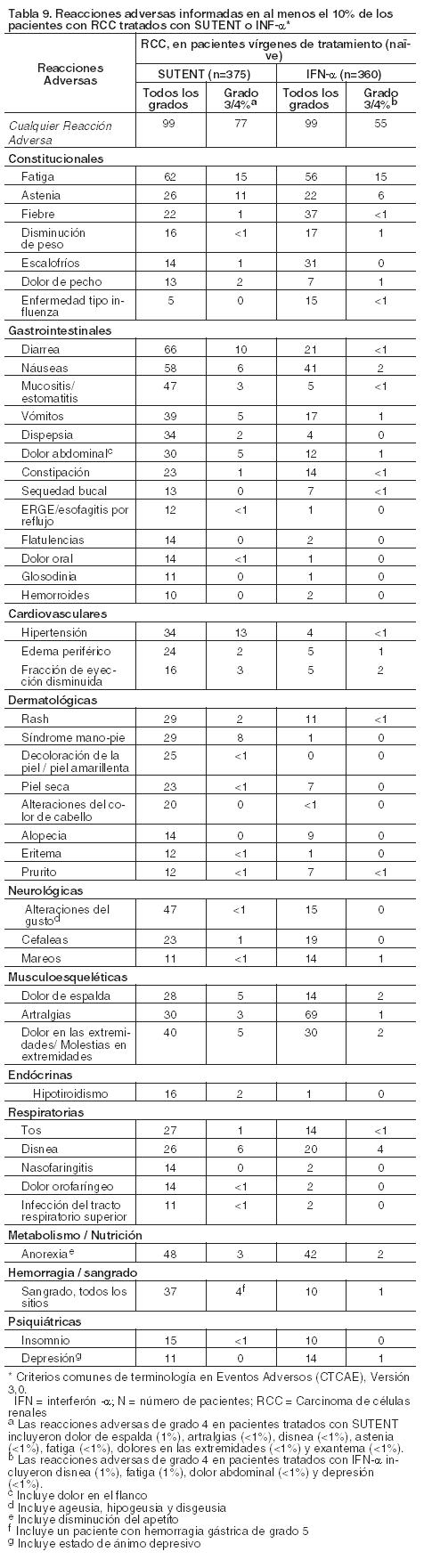
Las anormalidades de laboratorio de grado 3 y 4 emergentes del tratamiento se presentan en la tabla 8.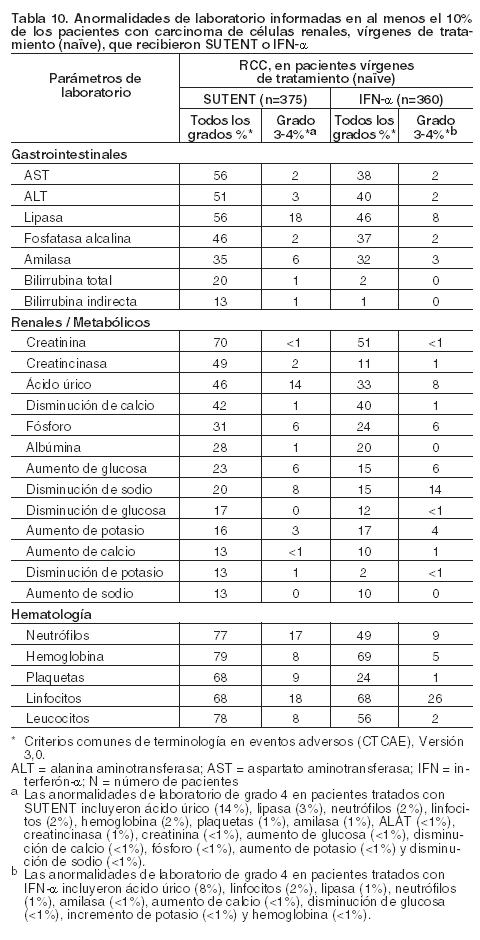
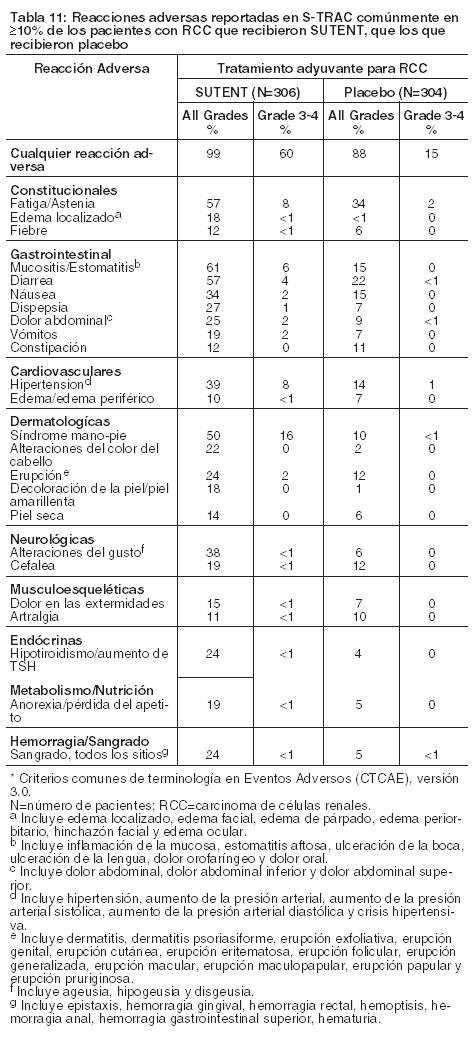
Seguridad a largo plazo en RCC: La seguridad a largo plazo de SUTENT en pacientes con RCC metastásico se analizó en 9 estudios clínicos completos realizados en entornos de tratamiento de primera línea, refractario a bevacizumab y refractario a citocina. El análisis incluyó a 5739 pacientes, de los cuales 807 (14%) fueron tratados durante al menos 2 años y 365 (6%) durante al menos 3 años. El tratamiento prolongado con SUTENT no pareció estar asociado con nuevos tipos de reacciones adversas. No hubo un aumento aparente en la incidencia anual de reacciones adversas, en puntos de tiempo posteriores. El hipotiroidismo aumentó durante el segundo año de tratamiento con nuevos casos informados hasta el año 4. Tratamiento adyuvante para RCC: La seguridad de SUTENT se evaluó en un ensayo aleatorizado, doble ciego, controlado con placebo; S-TRAC; en el que los pacientes que se habían sometido a una nefrectomía por RCC recibieron SUTENT 50 mg al día (n=306) en el esquema 4/2 o placebo (n=304). La duración media del tratamiento fue 12,4 meses (rango: 0,13-14,9) para SUTENT y 12,4 meses (rango: 0,03-13,7) para placebo. La interrupción permanente debido a una reacción adversa ocurrió en el 28% de los pacientes con SUTENT y el 6% con