DUXETIN® 30 - 60
GADOR
Antidepresivo.
Composición.
Cada cápsula con microgránulos con recubrimiento entérico de DUXETIN® 30 contiene: Duloxetina base (como clorhidrato 33,70 mg) 30 mg. Excipientes c.s.: Sacarosa, Esferas de Azúcar, Talco, Hipromelosa, Acetato Succinato de Hipromelosa, Dióxido de titanio y Citrato de Trietilo. Las cápsulas de gelatina contienen, además de ésta, Índigo Carmín y Dióxido de Titanio. Cada cápsula con microgránulos con recubrimiento entérico de DUXETIN® 60 contiene: Duloxetina base (como clorhidrato 67,40 mg) 60 mg. Excipientes c.s.: Sacarosa, Esferas de Azúcar, Talco, Hipromelosa, Acetato Succinato de Hipromelosa, Dióxido de titanio y Citrato de Trietilo. Las cápsulas de gelatina contienen, además de ésta, Índigo Carmín, Óxido de hierro amarillo y Dióxido de Titanio. Los microgránulos están diseñados para prevenir la degradación del fármaco en el medio ácido del estómago.
Farmacología.
Descripción: DUXETIN® (cápsulas con microgránulos con recubrimiento entérico) es un potente inhibidor de la recaptación neuronal de serotonina y nor epinefrina (ISRSN) de administración oral. Su denominación química es clorhidrato de (+)-(S)-N-metil-c-(1-naftiloxi)-2-tiofenopropilamina. La fórmula empírica es C18H19NOS·HCl, que corresponde a un peso molecular de 333,88. La fórmula estructural es:
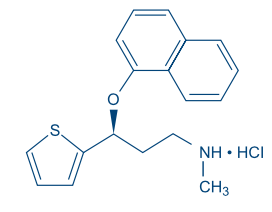
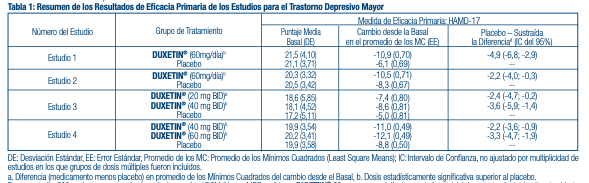
El clorhidrato de duloxetina es un sólido de un color que va de blanco a blanco ligeramente pardo, y que es ligeramente soluble en agua. Mecanismo de acción: Aunque los mecanismos exactos de los antidepresivos, las acciones centrales inhibidoras de dolor y ansiolíticas de la duloxetina en humanos aún no se conocen, se cree que estas acciones están relacionadas con su potenciación de actividad serotoninérgica y noradrenérgica en el SNC. Farmacodinámica: Estudios preclínicos han demostrado que la duloxetina es un potente inhibidor de la recaptación de serotonina y norepinefrina neuronal, y un inhibidor débil de la recaptación de dopamina. La duloxetina no tiene afi nidad signifi cativa por los receptores dopaminérgicos, adrenérgicos, colinérgicos, histaminérgicos, opioides, de glutamato y GABA in vitro. La duloxetina no inhibe la monoaminooxidasa (MAO). DUXETIN® se encuentra dentro de una clase de fármacos conocidos por afectar la resistencia uretral. Si se desarrollan síntomas de vacilación urinaria durante el tratamiento con DUXETIN®, debe considerarse la posibilidad de que ellos pueden estar relacionados con el fármaco. Farmacocinética: La duloxetina tiene una vida media de eliminación de alrededor 12 horas (rango entre 8 y 17 horas) y su farmacocinética es proporcional a la dosis en el rango terapéutico. Por lo general, las concentraciones plasmáticas en el equilibrio (Steady - State) se logran luego de 3 días de dosifi cación. La eliminación de la duloxetina ocurre principalmente a través del metabolismo hepático que involucra a dos isoenzimas del Citocromo P450: la CYP2D6 y la CYP1A2. Absorción y Distribución: Se logra una buena absorción del clorhidrato de duloxetina administrado por vía oral. Existe un lapso de tiempo de 2 horas y media hasta que empieza la absorción (lapso de T), las concentraciones plasmáticas máximas (Cmax) de duloxetina ocurrirán 6 horas después de la dosis. Los alimentos no afectan la Cmax de duloxetina, pero retrasan el tiempo que lleva alcanzar las concentraciones pico de 6 a 10 horas y, disminuyen ligeramente la extensión de la absorción (ABC) en aproximadamente 10%. Existe un retraso de 3 horas en la absorción y un incremento de un tercio en la depuración aparente de la duloxetina durante la noche en comparación con la mañana. El volumen de distribución aparente promedio es de alrededor 1640 L. La duloxetina está altamente unida ( > 90%) a las proteínas en el plasma humano, uniéndose principalmente a la albúmina y a la glicoproteína acídica a-1. La unión de la duloxetina a las proteínas plasmáticas no se ve afectada por el deterioro renal o hepático. Metabolismo y Eliminación: La Biotransformación y la disposición de la duloxetina en humanos han sido determinadas después de la administración oral de la duloxetina marcada con 14C. La duloxetina constituye cerca del 3% del material total radiomarcado en el plasma, indicando que ésta experimenta un extenso metabolismo hacia numerosos metabolitos. Las principales vías de biotransformación de la duloxetina incluyen la oxidación del anillo naftilo, seguido de una conjugación y posterior oxidación. Tanto la CYP2D6 como la CYP1A2 catalizan la oxidación del anillo de naftilo in vitro. Los 2 principales metabolitos encontrados en el plasma y orina son el glucurónido conjugado de 4-hidroxi duloxetina y el sulfato conjugado de 5-hidroxi, 6-metoxi duloxetina. Muchos metabolitos adicionales han sido identifi cados en la orina, algunos de ellos sólo representaban vías menores de eliminación. Los principales metabolitos de circulación no son farmacológicamente activos. En la orina sólo se encuentran trazas ( < 1% de la dosis) de duloxetina sin cambio. La mayor parte (alrededor del 70%) de la dosis de duloxetina aparece en la orina como metabolitos de la duloxetina; cerca del 20% es excretado en las heces. Toxicología preclínica: Carcinogénesis, Mutagénesis, Daño a la Fertilidad: Carcinogénesis: Se administró duloxetina en la dieta de ratas y ratones durante 2 años. En los ratones hembra que recibieron duloxetina en una dosis de 140 mg/kg/día (6 veces la dosis máxima recomendada para humanos (DMRH) de 120 mg/día sobre una base de mg/m2, hubo un incremento en la incidencia de adenomas hepatocelulares y carcinomas. La dosis para la que no se verifi caron efectos fue 50 mg/kg/día (2 veces la DMRH). La incidencia de tumores no aumentó en los ratones macho que recibieron duloxetina en dosis de hasta 100 mg/kg/día (4 veces la DMRH). En el caso de las ratas, las dosis dietéticas de duloxetina de hasta 27 mg/kg/día en hembras (2 veces la DMRH) y hasta 36 mg/kg/día en machos (3 veces la DMRH) no aumentó la incidencia de tumores. Mutagénesis: La duloxetina no demostró potencial mutagénico en un ensayo de mutación bacteriana reversa in vitro (prueba Ames) y no demostró ser clastogénica en una prueba de aberración cromosómica in vivo realizada a células de la médula ósea de ratones. Además, no resultó genotóxica en un ensayo in vitro de mutación genética mamífera en células de linfoma de ratón o en un ensayo de síntesis de ADN in vitro no programado en hepatocitos de rata, ni tampoco indujo el intercambio de cromátidas hermanas en la médula ósea de un hámster chino in vivo. Daño a la fertilidad: La administración oral de duloxetina a ratas hembra o macho antes y durante el apareamiento en dosis de hasta 45 mg/kg/día (4 veces la DMRH) no alteró el apareamiento o la fertilidad. Estudios clínicos: Se ha establecido la efi cacia de DUXETIN® en los siguientes estudios adecuados y bien controlados: Trastorno Depresivo Mayor (MDD por sus siglas en inglés): 4 estudios de corta duración y 1 estudio de mantenimiento en adultos [véase Estudios Clínicos]. Trastorno de Ansiedad Generalizada (GAD por sus siglas en inglés): 3 estudios de corta duración en adultos y 1 estudio de mantenimiento en adultos [véase Estudios Clínicos]. Dolor Neuropático Periférico de origen Diabético (DPNP por sus siglas en inglés): Dos estudios de 12 semanas en adultos [véase Estudios Clínicos]. Fibromialgia (FM): Dos estudios en adultos (uno con 3 meses de duración y el otro con 6 meses de duración) [véase Estudios Clínicos]. Dolor Musculoesquelético Crónico: Dos estudios de 12 a 13 semanas en pacientes adultos con dolor crónico en la zona lumbar (CLBP por sus siglas en inglés) y un estudio de 13 semanas en pacientes adultos con dolor crónico causado por la osteoartritis [véase Estudios Clínicos]. Trastorno Depresivo Mayor: La efi cacia de DUXETIN® como tratamiento para la depresión se determinó en 4 estudios aleatorios, doble ciego, controlados por placebo, de dosis fi jas en pacientes adultos ambulatorios (18-83 años) que reunieran los criterios del Manual DSM-IV para la depresión mayor. En 2 estudios, los pacientes fueron seleccionados al azar para recibir DUXETIN® 60 mg una vez al día (n=123 y n=128, respectivamente) o placebo (n=122 y n=139, respectivamente) durante 9 semanas; en el tercer estudio, los pacientes fueron seleccionados al azar para recibir DUXETIN® 20 o 40 mg dos veces al día (n=86 y n=91, respectivamente) o placebo (n=89) durante 8 semanas; en el cuarto estudio, los pacientes fueron seleccionados al azar para recibir DUXETIN® 40 o 60 mg dos veces al día (n=95 y n=93, respectivamente) o placebo (n=93) durante 8 semanas. No existe evidencia de que las dosis mayores a 60 mg/día ofrezcan más benefi cios. En los 4 estudios, DUXETIN® demostró superioridad sobre el placebo según se midió en el puntaje total de la Escala de Depresión de Hamilton de 17 ítems (HAMD-17). (Estudios 1-4 en la Tabla 1). En todos estos estudios clínicos, los análisis de la relación entre el resultado del tratamiento y la edad, el género y la raza no sugirieron ninguna respuesta diferencial basadas en estas características de los pacientes.
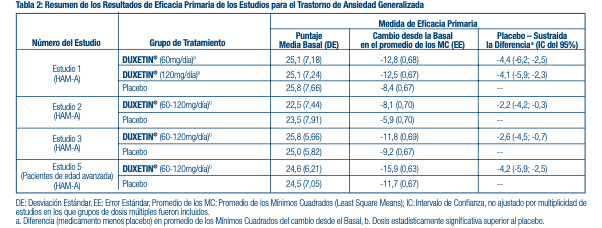
En otro estudio, 533 pacientes que reunían los criterios según el DSM-IV para MDD recibieron DUXETIN® 60 mg una vez al día durante la fase inicial de un tratamiento de etiqueta abierta de 12 semanas. Doscientos setenta y ocho pacientes que respondieron al tratamiento de etiqueta abierta (defi nidos como quienes reunieron los siguientes criterios durante las semanas 10 y 12: puntaje fi nal del HAMD-17 -9, Impresiones clínicas globales de severidad (CGI-S) -2, y quienes no reunieron los criterios del DSM-IV para MDD) fueron asignados al azar para continuar con DUXETIN® en la misma dosis (n=136) o el placebo (n=142) durante 6 meses. Los pacientes que recibieron DUXETIN® experimentaron un tiempo estadísticamente signifi cativo más prolongado para sufrir una recaída de depresión que los pacientes que recibían el placebo. La recaída se defi nió como un aumento del resultado CGI-S de 2 puntos en comparación con el resultado obtenido en la semana 12, así como quienes reunieron los criterios del DSM-IV para MDD en 2 visitas consecutivas en un lapso de por lo menos 2 semanas, para el cual el criterio temporal de 2 semanas solo debía cumplirse en la segunda visita. La efectividad de DUXETIN® en pacientes hospitalizados con trastorno depresivo mayor no ha sido estudiada. Trastorno de Ansiedad Generalizada: La efi cacia de DUXETIN® en el tratamiento del trastorno de ansiedad generalizada se estableció en 1 ensayo aleatorio, doble ciego, controlado por placebo, de dosis fi ja y 2 ensayos aleatorios, doble ciego, controlado por placebo, de dosis fl exible en pacientes adultos ambulatorios entre 18 y 83 años de edad que reunieron los criterios del DSM-IV para GAD. En un estudio de dosis fl exible y en el estudio de dosis fi ja, la dosis inicial fue de 60 mg una vez al día en que se permitió la titulación hasta 30 mg una vez al día por razones de tolerabilidad antes de aumentar a 60 mg una vez al día. El quince por ciento de los pacientes tuvieron sus dosis tituladas. Un estudio de dosis fl exible tenía una dosis inicial de 30 mg una vez al día durante 1 semana antes de aumentarla a 60 mg una vez al día. Los 2 estudios de dosis fl exibles tuvieron titulación de dosis de DUXETIN® con dosis que fueron desde 60 mg una vez al día para 120 mg una vez al día (N=168 y N=162) en comparación con placebo (N=159 y N=161) durante un periodo de tratamiento de 10 semanas. La dosis media para los pacientes que completaron el estudio hasta el punto fi nal de los estudios de dosis fl exibles fue 104,75 mg/día. El estudio de dosis fi ja evaluó dosis de DUXETIN® de 60 mg una vez al día (N=168) y de 120 mg una vez al día (N=170) en comparación con placebo (N=175) durante 9 semanas de tratamiento. Si bien se ha demostrado la efi cacia de una dosis de 120 mg/día, no existe evidencia de que las dosis mayores a 60 mg/día ofrezcan benefi cios adicionales. En los 3 estudios, DUXETIN® demostró superioridad sobre el placebo de acuerdo al puntaje total de la medición de la Escala de Ansiedad de Hamilton (HAM-A) (Estudios 1-3 en la Tabla 2) y al resultado global de insufi ciencia funcional de la Escala de Discapacidad de Sheehan (SDS). La SDS es una medición compuesta del grado en el que los síntomas emocionales perturban el funcionamiento del paciente en 3 campos de la vida: trabajo/estudios, vida social/actividades placenteras, y vida familiar/responsabilidades en el hogar. En otro estudio, 887 pacientes que reunieron los criterios de DSM-IV-TR para GAD recibieron DUXETIN® 60 mg a 120 mg una vez al día durante la fase inicial del tratamiento de etiqueta abierta de 26 semanas. Cuatrocientos veintinueve pacientes que respondieron al tratamiento de etiqueta abierta (defi nidos como quienes reunieron los siguientes criterios en las semanas 24 y 26: una disminución del resultado total inicial de la HAM-A en por lo menos 50% a un puntaje no mayor de 11, y un puntaje de 1 o 2 en las Impresiones Clínicas Globales de Mejora [CGI-Mejora]) fueron asignados al azar para continuar con DUXETIN® en la misma dosis (n=216) o con el placebo (n=213) y eran observados ante posibles recaídas. De los pacientes seleccionados al azar, un 73% tenía un estatus de receptor por al menos 10 semanas. La recaída se defi nió como el aumento del resultado de las CGI-Severidad en al menos 2 puntos a un puntaje de 4 y un diagnóstico MINI (Mini Entrevista Internacional Neuropsiquiátrica) de GAD (excluyendo la duración), o discontinuación debido a la falta de efi cacia. Los pacientes que tomaron DUXETIN® experimentaron un tiempo estadísticamente signifi cativo más prolongado para sufrir una recaída de GAD que los pacientes que recibieron el placebo. Los análisis del subgrupo no indicaban que hubiese diferencias en los resultados del tratamiento como una función de la edad o el género. La efi cacia de DUXETIN® en el tratamiento de pacientes de 65 años de edad con trastorno de ansiedad generalizada se estableció en un estudio de 10 semanas, de dosis fl exible, aleatorio, doble ciego, controlado por placebo, en adultos de 65 años de edad que reunieron los criterios de DSM-IV-TR para GAD. En este estudio la dosis inicial fue de 30 mg una vez al día durante 2 semanas antes de que se les permitiera aumentos de dosis en incrementos de 30 mg en las semanas de tratamiento 2, 4 y 7, hasta 120 mg basado en el juicio del investigador de la respuesta clínica y tolerabilidad. La dosis media para los pacientes que completaron la fase del tratamiento agudo de 10 semanas fue 50,95 mg. Los pacientes tratados con DUXETIN® (N=151) demostraron una mejoría signifi cativamente mayor en comparación con el placebo (N=140) en el promedio del cambio desde el basal hasta el punto fi nal, según lo medido por el puntaje total de la medición de la Escala de Ansiedad de Hamilton (HAM-A) (Estudio 5 en la Tabla 2).
Dolor Neuropático Periférico de origen Diabético: La efi cacia de DUXETIN® para el tratamiento del dolor neuropático asociado a la neuropatía diabética periférica fue establecida en 2 estudios de 12 semanas, aleatorios, de doble ciego, controlados por placebo, de dosis fi ja, en pacientes adultos con dolor neuropático periférico de origen diabético durante al menos 6 meses. El estudio DPNP-1 y el estudio DPNP-2 enrolaron un total de 791 pacientes de los cuales 592 (75%) completaron los estudios. Los pacientes enrolados tenían diabetes mellitus de tipo I y II con un diagnóstico de polineuropatía sensomotriz simétrica distal dolorosa durante al menos 6 meses. Los pacientes tuvieron un puntaje inicial de dolor ≥ 4 sobre una escala de 11 puntos que iba de 0 (sin dolor) a 10 (peor dolor posible). A los pacientes se les permitió tomar hasta 4 g de acetaminofén por día según era necesario para calmar el dolor, además de DUXETIN®. Los pacientes registraban su dolor diario en un cuaderno de notas. Ambos estudios compararon DUXETIN® 60 mg una vez al día o 60 mg dos veces al día con un placebo. El DPNP-1 además comparó DUXETIN® 20 mg con un placebo. Un total de 457 pacientes (342 en DUXETIN®, 115 en placebo) fueron enrolados en el DPNP-1 y un total de 334 pacientes (226 en DUXETIN® y 108 en placebo) fueron enrolados en el DPNP-2. El tratamiento con DUXETIN® 60 mg una o dos veces al día mejoró estadística y signifi cativamente los puntajes fi nales promedio de dolor desde el inicio y aumentó la proporción de pacientes con por lo menos un 50% de reducción de los puntajes de dolor desde el inicio. Fibromialgia: La efi cacia de DUXETIN® en el tratamiento de la fi bromialgia se estableció en dos estudios aleatorios, de doble ciego, controlados por placebo, de dosis fi ja, en pacientes adultos que reunieron los criterios del Colegio Americano de Reumatología para la fi bromialgia (un historial de dolor generalizado de 3 meses, y dolor presente en 11 o más de los 18 puntos sensibles específi cos). El estudio FM-1 tuvo tres meses de duración y enroló solo a pacientes mujeres. El estudio FM-2 tuvo seis meses de duración y enroló a pacientes varones y mujeres. Aproximadamente un 25% de participantes tuvo un diagnóstico comórbido de trastorno depresivo mayor (MDD por sus siglas en inglés). El FM-1 y el FM-2 enrolaron un total de 874 pacientes de los cuales 541 (62%) completaron los estudios. Los pacientes tuvieron un puntaje inicial de dolor de 6.5 en una escala de 11 puntos que iba de 0 (sin dolor) a 10 (peor dolor posible). Ambos estudios compararon DUXETIN® 60 mg una vez al día o 120 mg diaria (administrada en dosis divididas en el FM-1 y como una sola dosis diaria en el FM-2) con el placebo. El FM-2 además comparó DUXETIN® 20 mg con el placebo durante los tres meses iniciales de un estudio de seis meses. Un total de 354 pacientes (234 con DUXETIN®, 120 con placebo) fueron enrolados en el FM-1 y un total de 520 pacientes (376 con DUXETIN®, 144 con placebo) fueron enrolados en el FM-2 (5% varones, 95% mujeres). El tratamiento con DUXETIN® 60 mg o 120 mg diaria estadística y signifi cativamente mejoró los puntajes fi nales promedio de dolor desde el punto inicial y aumentó la proporción de pacientes con al menos un 50% de reducción del puntaje del dolor en la etapa inicial. La reducción del dolor se observó en pacientes con y sin MDD comórbido. Sin embargo, el grado de reducción del dolor puede ser mayor en pacientes con MDD comórbido. Asimismo, el beneficio de incrementar la dosis en participantes no receptores de DUXETIN® con una dosis de 60 mg/día fue evaluado en un estudio aparte. Los pacientes fueron inicialmente tratados con DUXETIN® 60 mg una vez al día durante ocho semanas en un estudio de etiqueta abierta. Posteriormente, quienes completaron el estudio en esta fase fueron seleccionados al azar para recibir un tratamiento doble ciego con DUXETIN® con una dosis de 60 mg una vez al día o 120 mg una vez al día. Aquellos pacientes considerados como no receptores, donde la respuesta estaba defi nida en al menos un 30% de reducción del puntaje de dolor desde el inicio hasta el fi nal del tratamiento de 8 semanas, tenían pocas probabilidades de reunir los criterios de respuesta al fi nal de las 60 semanas de tratamiento si en el ciego se les ajustaba a DUXETIN® 120 mg en comparación con los que continuaban en el ciego con DUXETIN® con una dosis de 60 mg. Dolor Musculoesquelético Crónico: DUXETIN® está indicado para el tratamiento del dolor musculoesquelético crónico. Esto se estableció en estudios con pacientes con dolor crónico en la zona lumbar y dolor crónico causado por la osteoartritis. Estudios en el dolor crónico en la zona lumbar: La efi cacia de DUXETIN® en el dolor crónico en la zona lumbar (CLBP por sus siglas en inglés) fue evaluada en dos ensayos clínicos aleatorios, doble ciego, controlados por placebo de 13 semanas de duración (estudio CLBP-1 y estudio CLBP-2), y uno de 12 semanas de duración (CLBP-3). El CLBP-1 y el CLBP-3 demostraron la efi cacia de DUXETIN® en el tratamiento del dolor crónico en la zona lumbar. Los pacientes de todos los estudios no mostraron señales de radiculopatía o estenosis espinal. Estudios en dolor crónico causado por la osteoartritis: La efi cacia de DUXETIN® en el dolor crónico causado por la osteoartritis fue evaluada en 2 ensayos clínicos aleatorios, doble ciego, controlados por placebo de 13 semanas de duración (estudio OA-1 y estudio OA-2). Todos los pacientes de ambos estudios cumplieron con los criterios clínicos y radiográfi cos del ACR para la clasifi cación de la osteoartritis idiopática de la rodilla. La aleatorización fue estratifi cada por el estatus de uso inicial de AINE de los pacientes. Los pacientes asignados a DUXETIN® comenzaron el tratamiento en ambos estudios con una dosis de 30 mg una vez al día durante una semana. Después de la primera semana, la dosis de DUXETIN® aumentó a 60 mg una vez al día. Luego de 7 semanas de tratamiento con DUXETIN® 60 mg una vez al día, los pacientes del OA-1 con una respuesta sub óptima al tratamiento ( < 30% reducción del dolor) y que toleraron la duloxetina 60 mg una vez al día aumentaron su dosis a 120 mg. No obstante, en el OA-2, todos los pacientes, independientemente de su respuesta al tratamiento después de 7 semanas, fueron seleccionados al azar para continuar recibiendo DUXETIN® 60 mg una vez al día o incrementar su dosis a 120 mg una vez al día durante el tiempo restante del estudio. Los pacientes en los grupos tratados con placebo en ambos estudios recibieron un placebo igual para todo el tiempo que duraban los estudios. En el caso de ambos estudios, los análisis de efi cacia se realizaron usando datos de 13 semanas de los grupos de tratamiento combinado de DUXETIN® 60 mg y 120 mg una vez al día en comparación con el grupo placebo. En el estudio OA-1 después de 13 semanas de tratamiento, los pacientes que tomaban DUXETIN® mostraron una reducción del dolor signifi cativamente mayor. Los análisis de subgrupo no indicaron que se observaran diferencias en los resultados de tratamiento como resultado del uso de AINES. En el estudio OA-2 después de 13 semanas de tratamiento, los pacientes que tomaban DUXETIN® no mostraron una reducción del dolor signifi cativamente mayor.
Indicaciones.
DUXETIN® está indicado para el tratamiento del: Trastorno Depresivo Mayor (DSM IV) [véase Estudios Clínicos]. Trastorno de Ansiedad Generalizada (DSM IV) [véase Estudios Clínicos]. Neuropatía Periférica de origen Diabético [véase Estudio Clínicos]. Fibromialgia [véase Estudios Clínicos]. Dolor Musculoesquelético Crónico [véase Estudios Clínicos].
Dosificación.
Ingerir DUXETIN® entero. No masticar o triturar. No abrir la cápsula y esparcir su contenido sobre los alimentos o mezclarlo con líquidos. Todo esto puede afectar el recubrimiento entérico. DUXETIN® puede ser administrado independientemente de los alimentos. Si olvidó tomar una dosis de DUXETIN®, tome la dosis omitida tan pronto como se acuerde. Si es casi la hora para la próxima dosis, no tome la dosis omitida y tome la siguiente dosis a la hora habitual. No tome dos dosis de DUXETIN® a la vez. Dosis para el Tratamiento de Trastorno Depresivo Mayor: Administrar DUXETIN® en una dosis total de hasta 60 mg/día (ya sea una vez al día o 30 mg dos veces al día). Para algunos pacientes, puede ser conveniente comenzar con una dosis de 30 mg una vez al día por 1 semana, para permitir a los pacientes adaptarse al medicamento antes de aumentar la dosis a 60 mg una vez al día. Si bien se ha demostrado la efi cacia de una dosis de 120 mg/día, no existe evidencia de que las dosis mayores a 60 mg/día ofrezcan beneficios adicionales. No se ha evaluado de manera adecuada la seguridad de las dosis mayores a 120 mg/día. Reevaluar periódicamente para determinar la necesidad de tratamiento de mantenimiento y la dosis apropiada para dicho tratamiento [véase Estudios Clínicos]. Dosis para el Tratamiento de Trastorno de Ansiedad Generalizada: Para la mayoría de pacientes, iniciar con DUXETIN® 60 mg una vez al día. En el caso de otros pacientes, puede ser conveniente comenzar con 30 mg una vez al día durante 1 semana, para permitir a los pacientes adaptarse al medicamento antes de aumentar la dosis a 60 mg una vez al día. Si bien se ha demostrado la eficacia de una dosis de 120 mg/día, no existe evidencia de que las dosis mayores a 60 mg/día ofrezcan beneficios adicionales. Sin embargo, si se decide aumentar la dosis a más de 60 mg una vez al día, aumentar la dosis en incrementos de 30 mg una vez al día. No se ha evaluado de manera adecuada la seguridad de las dosis mayores a 120 mg/día. Reevaluar periódicamente para determinar la necesidad de seguir el tratamiento de mantenimiento y la dosis apropiada para dicho tratamiento [véase Estudios Clínicos]. Pacientes de edad avanzada: Iniciar DUXETIN® con una dosis de 30 mg una vez al día durante 2 semanas, antes de considerar un aumento a la dosis óptima de 60 mg. Posteriormente los pacientes pueden beneficiarse de dosis superiores a 60 mg una vez al día. Si se decide aumentar la dosis a más de 60 mg una vez al día, aumentar la dosis en incrementos de 30 mg una vez al día. La dosis máxima estudiada fue 120 mg/día. No se ha evaluado de manera adecuada la seguridad de las dosis mayores a 120 mg/día [véase Estudios Clínicos]. Dosis para el tratamiento de Dolor Neuropático Periférico de origen Diabético: Administrar DUXETIN® 60 mg una vez al día. No hay evidencia, sin embargo, de que las dosis superiores a 60 mg ofrezcan beneficios adicionales significativos y que la dosis más alta sea menos tolerada [véase Estudios Clínicos]. Se puede considerar una dosis inicial menor en el caso de pacientes para quienes la tolerabilidad es un tema de preocupación. Puesto que con frecuencia la diabetes se complica con enfermedades renales, considerar una dosis inicial baja y un aumento gradual de la misma en el caso de pacientes que sufren insufi ciencia renal [véase Dosificación y Modo de Administración, Propiedades Farmacológicas y Advertencias y Precauciones Especiales de Uso - Uso en Poblaciones Específicas]. Dosis para el tratamiento de Fibromialgia: Administrar DUXETIN® 60 mg una vez al día. Comenzar el tratamiento con una dosis de 30 mg una vez al día durante 1 semana, para permitir a los pacientes adaptarse al medicamento antes de aumentar la dosis a 60 mg una vez al día. Algunos pacientes pueden responder a la dosis inicial. No existe evidencia, sin embargo, de que las dosis mayores a 60 mg/día ofrezcan beneficios adicionales, aún en pacientes que no responden a las dosis de 60 mg. Las dosis mayores están asociadas a un mayor índice de reacciones adversas [véase Estudios Clínicos]. Dosis para el tratamiento de Dolor Musculoesquelético Crónico: Administrar DUXETIN® 60 mg una vez al día. Comenzar el tratamiento con una dosis de 30 mg por una semana, para permitir a los pacientes adaptarse al medicamento antes de aumentar la dosis a 60 mg una vez al día. No existe evidencia de que las dosis mayores ofrezcan beneficios adicionales, aún en pacientes que no responden a la dosis de 60 mg. Las dosis mayores están asociadas a un mayor índice de reacciones adversas [véase Estudios Clínicos]. Dosis en poblaciones especiales: Insuficiencia hepática - se recomienda no administrar DUXETIN® ordinariamente a pacientes con alguna insuficiencia hepática (ver Advertencias y Precauciones, Uso en poblaciones especificas). Insuficiencia renal severa - no se recomienda DUXETIN® en pacientes con una enfermedad renal o insuficiencia renal severa en etapa terminal (depuración de creatinina aproximada < 30 mL/min) (ver Uso en poblaciones especificas). Discontinuación de DUXETIN®: Reacciones adversas tras la discontinuación abrupta o gradual de DUXETIN® incluyen: mareos, dolor de cabeza, náusea, diarrea, parestesia, irritabilidad, vómitos, insomnio, ansiedad, hiperhidrosis, y fatiga. Se recomienda una reducción gradual en la dosifi cación en lugar de una suspensión abrupta cuando sea posible [véase Advertencias y Precauciones Especiales de Uso]. Paciente que cambia desde o hacia un Inhibidor de la Monoaminoxidasa (IMAO) que intenta tratar un Desorden Psiquiátrico: Deben transcurrir un mínimo de 14 días entre la discontinuación de un IMAO que intenta tratar un Desorden Psiquiátrico y el inicio de la terapia con DUXETIN®. En cambio, se deben procurar al menos 5 días posteriores a la suspensión de DUXETIN® antes de comenzar a tomar un IMAO que intenta tratar un Desorden Psiquiátrico [véase Contraindicaciones]. Uso de DUXETIN® con otros IMAOs tales como Linezolida o Azul de metileno: No inicie tratamiento con DUXETIN® en aquellos pacientes que están siendo tratados con linezolida o azul de metileno intravenoso porque existe un riesgo incrementado de síndrome serotoninérgico. Deben considerarse otras intervenciones, incluyendo hospitalización, en aquellos pacientes que requieren un tratamiento más urgente a una condición psiquiátrica [ver Contraindicaciones]. En algunos casos, un paciente que ya está recibiendo DUXETIN® podría requerir un tratamiento urgente con linezolida o azul de metileno intravenoso. Si no están disponibles alternativas aceptables para linezolida o azul de metileno intravenoso y los benefi cios potenciales del tratamiento con linezolida o azul de metileno intravenoso son ponderados para sopesar los riesgos de síndrome serotoninérgico en un paciente en particular, entonces DUXETIN® deberá suspenderse rápidamente para que la linezolida o el azul de metileno intravenoso puedan ser administrados. El paciente debe ser monitoreado para síntomas de síndrome serotoninérgico por 5 días o hasta 24 horas después de la última dosis de linezolida o azul de metileno intravenoso lo que ocurra primero. La terapia con DUXETIN® puede ser retomada 24 horas después de la última dosis de linezolida o azul de metileno intravenoso [ver Advertencias y Precauciones Especiales de Uso]. El riesgo de administrar azul de metileno por vías no intravenosas (tal como tabletas orales o inyección local) o en dosis intravenosa mucho menor a 1 mg/kg con DUXETIN® no está claro. El médico tratante debe, sin embargo, estar al tanto de la posibilidad de la aparición de síntomas del síndrome serotoninérgico con dicho uso [ver Advertencias y Precauciones Especiales de Uso].
Contraindicaciones.
Hipersensibilidad: Alergia a la duloxetina o a cualquiera de los componentes de la fórmula. Inhibidores de la monoaminoxidasa (IMAO): El uso de IMAOs (que intentan tratar desórdenes psiquiátricos) con DUXETIN® o dentro de los 5 días luego de haber detenido el tratamiento con DUXETIN® está contraindicado debido al riesgo incrementado de síndrome serotoninérgico. El uso de DUXETIN® dentro de los 14 días luego de haber detenido un IMAO que intenta tratar desórdenes psiquiátricos también está contraindicado [ver Dosifi cación y Modo de Administración y Advertencias y Precauciones Especiales de Uso]. Iniciar tratamiento con DUXETIN® en pacientes que están siendo tratados con IMAOs tales como linezolida o azul de metileno intravenoso también está contraindic ado debido al riesgo incrementado de síndrome serotoninérgico [ver Dosifi cación y Modo de Administración y Advertencias y Precauciones Especiales de Uso]. Glaucoma de ángulo estrecho no controlado: En los ensayos clínicos, DUXETIN® estuvo asociada a un incremento del riesgo de midriasis; por consiguiente, se debe evitar su uso en pacientes con glaucoma de ángulo estrecho no controlado [véase Advertencias y Precauciones Especiales de Uso]. Enfermedad hepática que produzca insufi ciencia hepática: Insuficiencia Renal severa (Aclaramiento o Depuración de Creatinina < 30 ml/min). Menores de 18 años de edad: Inhibidores potentes de la CYP1A2: DUXETIN® no debe utilizarse en combinación con fl uvoxamina, ciprofl oxacino o enoxacino dado que la combinación da lugar a concentraciones plasmáticas de duloxetina elevadas (ver Interacciones - Potencial de que DUXETIN® se vea afectada por otros fármacos). Hipertensión no controlada: El inicio del tratamiento con DUXETIN® está contraindicado en pacientes con hipertensión no controlada, ya que esta situación podría exponer a los pacientes a un riesgo potencial de crisis hipertensiva (ver Advertencias y Precauciones Especiales de Uso y Reacciones Adversas).
Reacciones adversas.
Las siguientes reacciones adversas graves están descritas abajo y en otras secciones del prospecto: Pensamientos y Comportamientos Suicidas en Adolescentes y Jóvenes Adultos [véase Advertencias y Precauciones Especiales de Uso]. Hepatotoxicidad [véase Advertencias y Precauciones Especiales de Uso]. Hipotensión Ortostática, Caídas y Síncope [véase Advertencias y Precauciones Especiales de Uso]. Síndrome Serotoninérgico [véase Advertencias y Precauciones Especiales de Uso]. Sangrado Anormal [véase Advertencias y Precauciones Especiales de Uso]. Reacciones Dermatológicas Severas [véase Advertencias y Precauciones Especiales de Uso]. Discontinuación del tratamiento con DUXETIN® [véase Advertencias y Precauciones Especiales de Uso]. Activación de Manía/Hipomanía [véase Advertencias y Precauciones Especiales de Uso]. Glaucoma de Ángulo Estrecho [véase Advertencias y Precauciones Especiales de Uso]. Convulsiones [véase Advertencias y Precauciones Especiales de Uso]. Efectos sobre la Tensión Arterial [véase Advertencias y Precauciones Especiales de Uso]. Interacciones Medicamentosas Clínicamente Importantes [véase Advertencias y Precauciones Especiales de Uso]. Hiponatremia [véase Advertencias y Precauciones Especiales de Uso]. Retención y Vacilación Urinaria [véase Advertencias y Precauciones Especiales de Uso]. Fuentes de información de los ensayos clínicos: Ya que los ensayos clínicos se realizan bajo condiciones sumamente variantes, los índices de reacciones adversas observados en los ensayos clínicos de un fármaco no pueden compararse directamente con los índices de los ensayos clínicos de otro fármaco y pueden no refl ejar los índices observados en la práctica. Las frecuencias establecidas de las reacciones adversas representan la proporción de individuos que experimentaron, al menos una vez, una reacción adversa como producto del tratamiento del tipo enumerado. Se consideró que una reacción era producto del tratamiento si ocurría por primera vez o empeoraba al recibir la terapia después de la evaluación inicial. Las reacciones reportadas durante los estudios no fueron causadas necesariamente por la terapia, y las frecuencias no refl ejan la impresión del investigador (evaluación) de causalidad. Adultos: La información descrita a continuación refl eja la exposición a duloxetina en ensayos controlados por placebo para Trastorno Depresivo Mayor (N=3779), Trastorno de Ansiedad Generalizada (N=1018), OA (N=503), Dolor Crónico en la zona Lumbar (N=600), Dolor Neuropático Periférico de origen Diabético (N=906), y FM (N=1294). La población estudiada tenía entre 17 y 89 años de edad; 65,7%, 60,8%, 60,6%, 42,9%, y 94,4% mujeres; y 81,8%, 72,6%, 85,3 %, 74,0%, y 85,7% de raza blanca para Trastorno Depresivo Mayor, Trastorno de Ansiedad Generalizada, OA y Dolor Crónico en la zona Lumbar, Dolor Neuropático Periférico de origen Diabético, y FM, respectivamente. La mayoría de los pacientes recibió dosis totales de 60 a 120 mg diarias [véase Estudios Clínicos]. La información a continuación no incluye los resultados de los ensayos para la evaluación de la efi cacia de DUXETIN® en pacientes 65 años de edad para el tratamiento del Trastorno de Ansiedad Generalizada; sin embargo las reacciones adversas observadas en la muestra geriátrica, fueron comúnmente similares a las reacciones adversas en la populación adulta general. Reacciones Adversas Reportadas como Razones para Discontinuar el Tratamiento en Ensayos en Adultos Controlados por Placebo: Trastorno Depresivo Mayor: Aproximadamente un 8,4% (319/3779) de los pacientes que recibieron DUXETIN® en ensayos controlados por placebo para Trastorno Depresivo Mayor discontinuaron el tratamiento debido a una reacción adversa, en comparación con un 4,6% (117/2536) de los pacientes que recibieron placebo. Las náuseas (1,1% con DUXETIN®, 0,4% con placebo) fueron la única reacción adversa común reportada como razón para discontinuar el tratamiento y se consideró que estaba relacionada al fármaco (es decir, la discontinuación ocurrió en al menos un 1% de los pacientes tratados con DUXETIN® y a un índice de por lo menos el doble del placebo). Trastorno de Ansiedad Generalizada: Aproximadamente un 13,7% (139/1018) de los pacientes que recibieron DUXETIN® en los ensayos controlados por placebo para Trastorno de Ansiedad Generalizada discontinuaron el tratamiento debido a una reacción adversa, en comparación con un 5,0% (38/767) que recibió el placebo. Las reacciones adversas comunes reportadas como razón para discontinuar el tratamiento y que se consideraron que estaban relacionadas al fármaco (según defi nición anterior) incluyen náuseas (3,3% con DUXETIN®, 0,4% con placebo), mareo (1,3% con DUXETIN®, 0,4% con placebo). Dolor Neuropático Periférico de origen Diabético. Aproximadamente un 12,9% (117/906) de los pacientes que recibieron DUXETIN® en ensayos controlados por placebo para el Dolor Neuropático Periférico de origen Diabético discontinuó el tratamiento debido a una reacción adversa, en comparación con un 5,1% (23/448) que recibió el placebo. Las reacciones adversas comunes reportadas como razón para discontinuar el tratamiento y que se consideraron que estaban relacionadas al fármaco (según defi nición anterior) incluyen náuseas (3,5% con DUXETIN®, 0,7% con placebo), mareo (1,2% con DUXETIN®, 0,4% con placebo) y somnolencia (1,1% con DUXETIN®, 0,0% con placebo). Fibromialgia: Aproximadamente un 17,5% (227/1294) de los pacientes que recibió DUXETIN® en ensayos de 3 a 6 meses controlados por placebo para la FM discontinuó el tratamiento debido a una reacción adversa, en comparación con un 10,1% (96/955) que recibió placebo. Las reacciones adversas comunes reportadas como razón para discontinuar el tratamiento y que se consideraron que estaban relacionadas al fármaco (según defi nición anterior) incluyen náuseas (2,0% con DUXETIN®, 0,5% con placebo), cefalea (1,2% con DUXETIN®, 0,3% con placebo), somnolencia (1,1% con DUXETIN®, 0,0% con
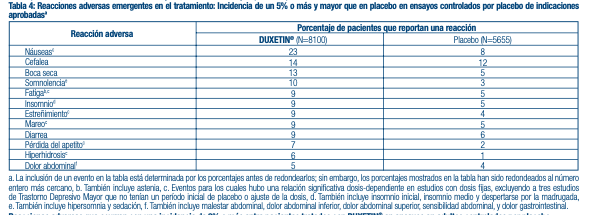
placebo), y fatiga (1,1% con DUXETIN®, 0,1% con placebo). Dolor Crónico causado por la Osteoartritis: Aproximadamente un 15.7% (79/503) de los pacientes que recibió DUXETIN® en ensayos de 13 semanas controlados por placebo para el dolor crónico causado por la OA discontinuó el tratamiento debido a una reacción adversa, en comparación con un 7.3% (37/508) que recibió placebo. Las reacciones adversas comunes reportadas como razón para discontinuar el tratamiento y que se consideraron que estaban relacionadas al fármaco (según defi nición anterior) incluyen náuseas (2.2% con DUXETIN®, 1.0% con placebo). Dolor Crónico en la Zona Lumbar: Aproximadamente un 16.5% (99/600) de los pacientes que recibió DUXETIN® en ensayos de 13 semanas controlados por placebo para el Dolor Crónico en la Zona Lumbar discontinuó el tratamiento debido a una reacción adversa, en comparación con un 6.3% (28/441) que recibió placebo. Las reacciones adversas comunes reportadas como razón para discontinuar el tratamiento y que se consideraron que estaban relacionadas al fármaco (según defi nición anterior) incluyen náuseas (3.0% con DUXETIN®, 0.7% con placebo), y somnolencia (1.0% con DUXETIN®, 0.0% con placebo). Reacciones Adversas Más Comunes en Adultos: Ensayos combinados para todas las indicaciones aprobadas. Las reacciones adversas más comunes observadas en pacientes tratados con DUXETIN® (incidencia de al menos un 5% y al menos el doble de la incidencia en pacientes que reciben placebo) fueron náuseas, boca seca, somnolencia, estreñimiento, pérdida de apetito, e hiperhidrosis. Dolor neuropático periférico de origen diabético: Las reacciones adversas más comunes observadas en pacientes tratados con DUXETIN® (según defi nición anterior) fueron náuseas, somnolencia, pérdida de apetito, estreñimiento, hiperhidrosis, y boca seca. Fibromialgia: Las reacciones adversas más comunes observadas en pacientes tratados con DUXETIN® (según defi nición anterior) fueron náuseas, boca seca, estreñimiento, somnolencia, pérdida de apetito, hiperhidrosis, y agitación. Dolor Crónico causado por la Osteoartritis: Las reacciones adversas más comunes observadas en pacientes tratados con DUXETIN® (según defi nición anterior) fueron náuseas, fatiga, estreñimiento, boca seca, insomnio, somnolencia y mareos. Dolor Crónico en la zona Lumbar: Las reacciones adversas más comunes observadas en pacientes tratados con DUXETIN® (según defi nición anterior) fueron náuseas, boca seca, insomnio, somnolencia, estreñimiento, mareo, y fatiga. Reacciones adversas que ocurren con una incidencia de 5% o más entre pacientes tratados con DUXETIN® en ensayos controlados por placebo: La Tabla 4 muestra la incidencia de las reacciones adversas que emergen durante el tratamiento en ensayos controlados por placebo ante indicaciones aprobadas que ocurrieron en un 5% de pacientes, o más, tratados con DUXETIN® y con una incidencia mayor que con el placebo.
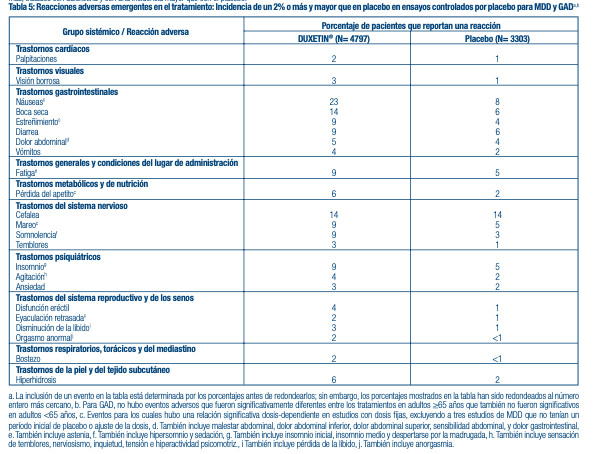
Reacciones adversas que ocurren con una incidencia de 2% o más entre pacientes tratados con DUXETIN® en ensayos en adultos controlados por placebo: Ensayos combinados para Trastorno Depresivo Mayor y Trastorno de Ansiedad Generalizada: La Tabla 5 presenta la incidencia de las reacciones adversas que emergen durante el tratamiento en ensayos controlados por placebo para Trastorno Depresivo Mayor y Trastorno de Ansiedad Generalizada ante indicaciones aprobadas que ocurrieron en un 2% de pacientes, o más, tratados con duloxetina y con una incidencia mayor que con el placebo.
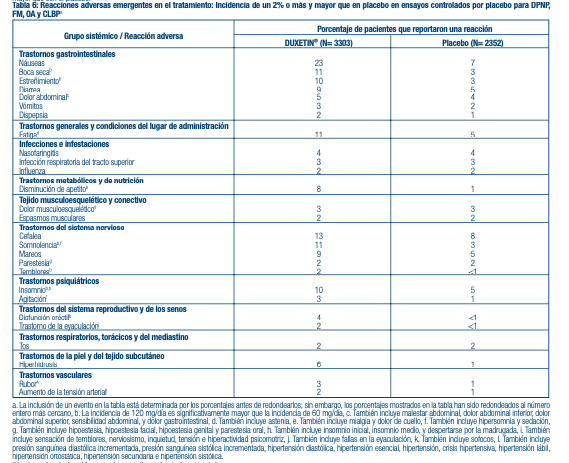
DPNP, FM, OA, y CLBP - La Tabla 6 presenta la incidencia de los eventos adversos que emergen durante el tratamiento que ocurrieron en un 2% de pacientes, o más, tratados con DUXETIN® (determinados antes del redondeo) en la fase aguda pre comercialización de DPNP, FM, OA y CLBP, y ensayos controlados por placebo de CLBP y con una incidencia mayor que con el placebo.
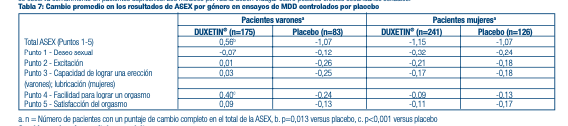
Efectos sobre la función sexual masculina y femenina en adultos: Los cambios en el deseo sexual, el rendimiento sexual y la satisfacción sexual a menudo ocurren como manifestaciones de trastornos psiquiátricos, pero también pueden ser producto de un tratamiento farmacológico. Ya que se presume que las reacciones adversas sexuales no son debidamente reportadas de forma voluntaria, la Escala de Experiencia Sexual de Arizona (ASEX), una medición validada diseñada para identifi car los efectos secundarios sexuales, fue usada de forma potencial en 4 ensayos de MDD controlados por placebo. En estos ensayos, tal como se muestra en la Tabla 7 a continuación, los pacientes tratados con DUXETIN® experimentaron de manera signifi cativa más disfunciones sexuales, según los resultados de la medición de ASEX, que los pacientes tratados con un placebo. El análisis de género mostró que esta diferencia ocurrió solo en varones. Los varones tratados con DUXETIN® experimentaron una mayor difi cultad para poder alcanzar el orgasmo (punto 4 de ASEX) que los varones tratados con un placebo. Las mujeres no experimentaron una mayor disfunción sexual cuando eran tratadas con DUXETIN® que con un placebo según el resultado fi nal de ASEX. Los números negativos signifi can una mejora del nivel inicial de la disfunción, la cual se observa comúnmente en pacientes depresivos. Los médicos por rutina deben indagar sobre posibles efectos secundarios sexuales.
Cambios en los signos vitales en adultos: En los ensayos clínicos por indicaciones aprobadas para realizar cambios del inicio al fi nal, el tratamiento con DUXETIN® estuvo asociado a los aumentos promedio de 0,23 mm Hg en la tensión arterial sístole y 0,73 mm Hg en la tensión arterial diástole en comparación con las disminuciones promedio de 1,09 mm HG sístole y 0,55 mm Hg diástole en pacientes tratados con un placebo. No hubo diferencia signifi cativa en la frecuencia de la tensión arterial alta sostenida (3 visitas consecutivas) [véase Advertencias y Precauciones Especiales de Uso]. El tratamiento con DUXETIN®, hasta 26 semanas en ensayos controlados por placebo por indicaciones aprobadas, producían normalmente un ligero aumento en la frecuencia cardíaca para cambiar del inicio al fi nal en comparación con el placebo hasta 1,37 latidos por minuto (incremento de 1,2 latidos por minuto en pacientes tratados con DUXETIN®, disminución de 0,17 latidos por minuto en pacientes tratados con placebo). Cambios en los resultados de análisis de laboratorio en adultos: El tratamiento con DUXETIN® en ensayos controlados por placebo para indicaciones aprobadas estuvo asociado a ligeros aumentos promedio desde el inicio hasta el fi nal en términos de ALT, AST, CPK, y fosfatasa alcalina; se observaron, además, valores infrecuentes, bajos, temporales, anormales para estos análisis en pacientes tratados con DUXETIN® al compararlos con pacientes tratados con un placebo [véase Advertencias y Precauciones Especiales de Uso]. Altos valores de bicarbonato, colesterol, y valores anormales (altos y bajos) de potasio fueron observados más frecuentemente en pacientes tratados con DUXETIN® que en los pacientes tratados con placebo. Cambios en el electrocardiograma en adultos: El efecto de DUXETIN® (160mg y 200mg administrados dos veces al día) sobre el estado estable fue evaluado en un estudio aleatorizado, doble ciego, de dos brazos en 117 mujeres sanas. No se detectó prolongación del intervalo QT. Parece ser que DUXETIN® está asociada con acortamiento, concentración dependiente pero no clínicamente signifi cativo, del intervalo QT. Otras reacciones adversas observadas durante la evaluación de ensayos clínicos antes y después de la comercialización de DUXETIN® en adultos: A continuación, se detalla una lista de las reacciones adversas emergentes durante el tratamiento, reportadas por los pacientes tratados con DUXETIN® en ensayos clínicos. En los ensayos clínicos de todas las indicaciones, 34,756 pacientes fueron tratados con DUXETIN®. De estos, un 26,9% (9337) tomaron DUXETIN® durante al menos 6 meses, y un 12,4% (4317) durante al menos un año. La siguiente lista no tiene como fi n incluir reacciones: (1) ya incluidas en tablas anteriores ni en indicaciones que fi guran en alguna otra parte del etiquetado, (2) donde el fármaco era una causa remota, (3) muy generales que casi no brindaban información, (4) que se consideraba tenían implicancias clínicas signifi cativas, o (5) que ocurrían a un índice igual o menor que con el placebo. Las reacciones están categorizadas según el sistema orgánico de acuerdo a las siguientes defi niciones: reacciones adversas frecuentes son aquéllas que ocurren en al menos 1/100 pacientes; reacciones adversas no frecuentes son aquéllas que ocurren en 1/100 a 1/1000 pacientes; reacciones esporádicas son aquéllas que ocurren en menos de 1/1000 pacientes. Trastornos cardíacos: Frecuente: palpitaciones; Infrecuente: infarto del miocardio y taquicardia. Trastornos del oído y laberinto: Frecuente: vértigo; Infrecuente: dolor de oído y tinnitus. Trastornos endocrinos: Infrecuente: hipotiroidismo. Trastornos de la visión: Frecuente: visión borrosa; Infrecuente: diplopia, ojo seco, y difi cultad visual. Trastornos gastrointestinales: Frecuente: fl atulencia; Infrecuente: disfagia, eructos, gastritis, hemorragia gastrointestinal, halitosis, y estomatitis; Raro: úlcera gástrica. Trastornos generales y condiciones del lugar de administración: Frecuente: escalofríos; Infrecuente: caídas, sensación de anormalidad, sensación de calor y/o frío, malestar general, y sed; Raro: trastornos en la marcha. Infecciones e infestaciones: Infrecuente: gastroenteritis y laringitis. Investigaciones: Frecuente: aumento de peso, pérdida de peso; Infrecuente: aumento del colesterol en la sangre. Trastornos metabólicos y de nutrición: Infrecuente: deshidratación e hiperlipidemia; Raro: dislipidemia. Trastorno del tejido musculoesquelético y conectivo: Frecuente: dolor musculoesquelético; Infrecuente: rigidez muscular y contracción muscular. Trastornos del sistema nervioso: Frecuente: disgeusia, letargo, y parestesia/hipoestesia; Infrecuente: disturbios de atención, disquinesia, mioclonos, y sueño de calidad defi ciente; Raro: disartria. Trastornos psiquiátricos: Frecuente: sueños anormales y trastorno del sueño; Infrecuente: apatía, bruxismo, desorientación/estado de confusión, irritabilidad, cambios de estado de ánimo, e intento de suicidio; Raro: suicidio. Trastornos renales y urinarios: Frecuente: frecuencia urinaria; Infrecuente: disuria, urgencia para orinar, nicturia, poliuria, y olor anormal de la orina. Trastornos del aparato reproductor y de los senos: Frecuente: anorgasmia/orgasmos anormales; Infrecuente: síntomas menopáusicos, disfunción sexual y dolor testicular; Raro: desorden menstrual. Trastornos respiratorios, torácicos y del mediastino: Frecuente: bostezo, dolor orofaríngeo; Infrecuente: estrechez o sensación de opresión en la garganta. Trastornos dermatológicos y del tejido subcutáneo: Frecuente: prurito; Infrecuente: sudor frío, dermatitis de contacto, eritema, mayor tendencia a hematomas, sudoración nocturna, y reacción fotosensible; Raro: equimosis. Trastornos vasculares: Frecuente: sofocos; Infrecuente: enrojecimiento, hipotensión ortostática, y frío periférico. Reportes espontáneos post comercialización: Durante el uso posterior a la aprobación de DUXETIN® se identifi caron las siguientes reacciones adversas. Ya que estas reacciones fueron reportadas voluntariamente por una población de un tamaño incierto, no siempre es posible estimar de forma confi able su frecuencia o establecer una relación causal a la exposición al fármaco. Las reacciones adversas reportadas desde su introducción en el mercado temporalmente relacionadas a la terapia con DUXETIN® y que no han sido mencionadas en otros espacios incluyen: pancreatitis aguda, reacción anafi láctica, agresión y enojo (particularmente en la fase inicial del tratamiento o después de la discontinuación del tratamiento), edema angioneurótico, glaucoma de ángulo estrecho, vasculitis cutánea (a veces asociadas con compromiso sistémico), trastorno extrapiramidal, galactorrea, hemorragia ginecológica, alucinaciones, hiperglucemia, hiperprolactinemia, hipersensibilidad, crisis hipertensiva, colitis (microscópica o inespecífi ca), espasmo muscular, sarpullido, síndrome de las piernas inquietas, convulsiones luego de discontinuar el tratamiento, arritmia supraventricular, tinnitus (luego de discontinuar el tratamiento), trismo y urticaria.
Advertencias.
Antes de iniciar la terapia con un antidepresivo se deben investigar cuidadosamente los antecedentes psiquiátricos del paciente, incluyendo historia familiar y personal de suicidios y trastorno bipolar. Úsese sólo por indicación y bajo vigilancia médica. Pensamientos y conductas suicidas en Adolescentes y Adultos jóvenes: El uso de antidepresivos con indicación aprobada por ensayos clínicos controlados en adultos con trastorno depresivo mayor y otras condiciones psiquiátricas deberá establecerse en un marco terapéutico adecuado a cada paciente en particular. Esto incluye: a) que la indicación sea hecha por médicos que puedan monitorear rigurosamente la emergencia de cualquier signo de agravamiento o aumento de la ideación suicida, como así también cambios conductuales con síntomas del tipo de agitación; b) que se tengan en cuenta los resultados de los últimos ensayos clínicos controlados; c) que se considere que el benefi cio clínico debe justifi car el riesgo potencial. Se han reportado los siguientes síntomas en pacientes adultos que están siendo tratados con antidepresivos para el trastorno depresivo mayor, así como para otras indicaciones, psiquiátricas y no psiquiátricas, ansiedad, agitación, ataques de pánico, insomnio, irritabilidad, hostilidad, agresividad, impulsividad, acatisia (inquietud psicomotriz), hipomanía y manía. Si bien no se ha establecido un vínculo entre la aparición de dichos síntomas y el empeoramiento de la depresión y/o la aparición de impulsos suicidas, existe la preocupación de que dichos síntomas pueden ser considerados precursores de comportamientos suicidas emergentes. Las familias y los responsables del cuidado de los pacientes que están siendo tratados con antidepresivos para el trastorno depresivo mayor, u otras indicaciones psiquiátricas y no psiquiátricas, deben estar prevenidos sobre la necesidad de monitorear a los pacientes y estar atentos ante la emergencia de síntomas como agitación, irritabilidad, cambios inusuales en la conducta, y los demás síntomas antes descritos, así como la aparición de la tendencia al suicidio, y reportarlos de inmediato a los profesionales de la salud. Dicho monitoreo debe incluir la observación diaria por parte de las familias y los responsables del cuidado de los pacientes. Si se ha decidido discontinuar el tratamiento, el medicamento debe ser reducido gradualmente tan pronto como sea posible, pero teniendo en cuenta que la discontinuación puede estar asociada a ciertos síntomas (véase Dosifi cación y Modo de Administración y Advertencias y Precauciones Especiales de Uso para conocer las descripciones de los riesgos de la discontinuación de DUXETIN®). La seguridad y efi cacia de DUXETIN® no ha sido establecida en pacientes pediátricos menores de 18 años y su uso no está dirigido a este grupo etario. (Ver Contraindicaciones y Dosifi cación y Modo de Administración - Dosis en poblaciones especiales). Los pacientes con trastorno depresivo mayor, adultos y pediátricos, pueden experimentar un empeoramiento de su depresión y/o la aparición de pensamientos y conductas suicidas (tendencia al suicidio) o cambios inusuales en la conducta, ya sea que estén tomando medicamentos antidepresivos o no, y este riesgo podría persistir hasta que ocurran remisiones de importancia. Se sabe que el suicidio es un riesgo de la depresión y algunos otros trastornos psiquiátricos, y estos trastornos son los principales factores pronóstico del suicidio. Sin embargo, por mucho tiempo ha habido gran preocupación respecto a si los antidepresivos pueden tener un papel de inducción del empeoramiento de la depresión y la emergencia de la tendencia al suicidio en ciertos pacientes durante la etapa inicial del tratamiento. Los análisis combinados de ensayos a corto plazo controlados por placebo de fármacos antidepresivos (ISRS y otros) mostraron que estos fármacos elevan el riesgo de pensamientos y conductas suicidas (tendencia al suicidio) en niños, adolescentes y jóvenes adultos (entre 18 y 24 años) con trastorno depresivo mayor y otros trastornos psiquiátricos. Los estudios a corto plazo no demostraron un incremento en el riesgo de la tendencia al suicidio con el uso de antidepresivos en comparación con el placebo en adultos mayores de 24 años, mientras que hubo una reducción del riesgo con el uso de antidepresivos en comparación con el placebo en adultos mayores de 65 años. Los análisis combinados de ensayos controlados por placebo en niños y adolescentes que sufren Trastorno Depresivo Mayor, trastorno obsesivo compulsivo (TOC), u otros trastornos psiquiátricos incluyeron un total de 24 ensayos a corto plazo de 9 fármacos antidepresivos en más de 4400 pacientes. Los análisis combinados de ensayos controlados por placebo en adultos con Trastorno Depresivo Mayor u otros trastornos psiquiátricos incluyeron un total de 295 ensayos a corto plazo (duración promedio de 2 meses) de 11 fármacos antidepresivos en más de 77,000 pacientes. Hubo una variación considerable en el riesgo de la tendencia al suicidio entre los fármacos, aunque una tendencia al incremento en pacientes jóvenes en el caso de casi todos los fármacos estudiados. Hubo diferencias en el riesgo absoluto de la tendencia al suicidio entre las difere ntes indicaciones, con una mayor incidencia en el caso del Trastorno Depresivo Mayor. El riesgo de las diferencias (fármaco vs. placebo), sin embargo, fue relativamente estable dentro del grupo etario y entre las indicaciones. Estas diferencias en el riesgo (diferencia fármaco - placebo en el número de casos de tendencia al suicidio por 1000 pacientes tratados) son presentadas en la Tabla 3.

No se reportaron suicidios en ninguno de los ensayos pediátricos. Sí hubo, sin embargo, suicidios en los ensayos con adultos, pero el número no fue sufi ciente para llegar a alguna conclusión sobre el efecto del fármaco respecto al suicidio. Se desconoce si el riesgo de la tendencia al suicidio se extiende a su uso a largo plazo, es decir, después de varios meses. Sin embargo, hay evidencia importante en los ensayos de mantenimiento controlados por placebo en adultos con depresión respecto a que el uso de antidepresivos puede retrasar la recurrencia de la depresión. Todos los pacientes tratados con antidepresivos por alguna indicación deben ser monitoreados de forma apropiada y observados muy de cerca para estar atentos al empeoramiento clínico, la tendencia al suicidio y cambios inusuales en la conducta, especialmente durante los primeros meses de la terapia con el fármaco, o cuando haya ajustes en la dosis, ya sea un incremento o una reducción. Debe considerarse el cambio del régimen terapéutico, incluyendo la posible discontinuación del medicamento, en pacientes cuya depresión es persistentemente peor, o que están experimentando una tendencia emergente al suicidio o síntomas que pueden ser precursores de un empeoramiento de la depresión o una tendencia al suicidio, especialmente cuando estos síntomas son severos, abruptos al inicio, o no eran parte de los síntomas usuales del paciente. Las prescripciones entregadas de DUXETIN® deben ser por el menor número de cápsulas que sea consistente con el buen tratamiento del paciente, a fi n de reducir el riesgo de una sobredosis. Evaluación de pacientes para la identifi cación del trastorno bipolar. Un episodio de depresión mayor puede ser la manifestación inicial de un trastorno bipolar. Generalmente se cree (aunque no se ha establecido en ensayos controlados) que tratar tales episodios solo con un antidepresivo puede aumentar la posibilidad de que se precipite un episodio maníaco o mixto en pacientes con riesgo de trastorno bipolar. Se desconoce si alguno de los síntomas antes descritos representa esa conversión. Sin embargo, antes de iniciar el tratamiento con un antidepresivo, los pacientes con síntomas depresivos deben ser debidamente observados para determinar si se encuentran en riesgo de un trastorno bipolar; dicha observación debe incluir el historial psiquiátrico detallado, incluyendo el historial familiar de suicidio, trastorno bipolar y depresión. Se debe tomar en cuenta que DUXETIN® no ha sido aprobado para su uso en el tratamiento de la depresión bipolar. Hepatotoxicidad: Ha habido reportes de insufi ciencia hepática, a veces fatal, en pacientes tratados con DUXETIN®. Estos casos se han presentado como hepatitis con dolor abdominal, hepatomegalia, y elevación de los niveles de transaminasa a más de veinte veces el límite superior de lo normal con o sin ictericia, refl ejando una tendencia combinada o hepatocelular del daño hepático. Se debe discontinuar DUXETIN® en pacientes que desarrollan ictericia u otra evidencia de disfunciones hepáticas clínicamente signifi cativas y no se debe reanudar su uso a menos que se haya establecido otra causa. También se han reportado casos de ictericia colestática con una elevación mínima de los niveles de transaminasa. Otros reportes posteriores a la comercialización indican que en pacientes con enfermedades hepáticas crónicas o cirrosis se observó la elevación de las transaminasas, bilirrubina, y fosfatasa alcalina. DUXETIN® aumentó el riesgo de la elevación de los niveles de las transaminasas séricas en los ensayos clínicos del programa de desarrollo. Las elevaciones de las transaminasas hepáticas resultaron en la discontinuación de un 0.3% (92/34,756) de pacientes tratados con DUXETIN®. En la mayoría de pacientes, el tiempo promedio para la detección de la elevación de la transaminasa fue de aproximadamente dos meses. En los ensayos en adultos, controlados por placebo para cualquier indicación, en pacientes con valores normales y anormales ALT en el punto de inicio, se produjo un incremento de ALT de más de 3 veces el límite superior normal en 1.25% (144/11,496) de los pacientes tratados con DUXETIN® en comparación con un 0.45% (39/8716) de los pacientes tratados con placebo. En los estudios controlados por placebo usando un diseño de dosis fi ja, hubo evidencia de una relación de respuesta a la dosis respecto al aumento de los valores de ALT y AST de más de 3 veces el límite superior normal y de más de 5 veces el límite superior normal, respectivamente. Ya que es posible que DUXETIN® y el alcohol interactúen produciendo un daño hepático o que DUXETIN® agrave las enfermedades hepáticas pre-existentes, DUXETIN® no debe ser prescrito a pacientes que consuman alcohol de manera considerable o con evidencia de una enfermedad hepática crónica. Hipotensión ortostática, caídas y síncope: Se han reportado casos de hipotensión ortostática, caídas y síncope con dosis terapéuticas de DUXETIN®. El síncope y la hipotensión ortostática tienden a ocurrir dentro de la primera semana de terapia pero pueden también ocurrir en cualquier momento durante el tratamiento con DUXETIN®, especialmente después de incrementar las dosis. El riesgo de caída parece estar relacionado con el grado de disminución ortostática de la tensión arterial, así como otros factores que pueden aumentar el riesgo subyacente de caídas. En un análisis de los pacientes de todos los estudios controlados estudios, los pacientes tratados con DUXETIN® reportaron una mayor tasa de caídas en comparación con los pacientes tratados con placebo. El riesgo parece estar relacionado con la presencia disminución ortostática de la tensión arterial. El riesgo de disminución de la tensión arterial puede ser mayor en pacientes que toman simultáneamente medicamentos que inducen la hipotensión ortostática (tales como antihipertensivos) o son potentes inhibidores del CYP1A2 [véase Advertencias y Precauciones Especiales de Uso e Interacciones] y en pacientes que toman DUXETIN® en dosis superiores a 60 mg al día. Se debe considerar la disminución de la dosis o discontinuar DUXETIN® en pacientes que experimentan hipotensión ortostática sintomática, caídas y/o síncope durante la terapia con DUXETIN®. El riesgo de caída también pareció ser proporcional al riesgo subyacente de un paciente de caídas y pareció aumentar constantemente con la edad. Como los pacientes de edad avanzada tienden a tener un mayor riesgo subyacente a caídas debido a una mayor prevalencia de factores de riesgo tales como el uso de múltiples medicamentos, comorbilidad médica y trastornos de la marcha, el impacto del aumento de la edad por sí solo no está claro. Se han reportado caídas con consecuencias graves, como fracturas óseas y hospitalizaciones [véase Advertencias y Precauciones Especiales de Uso - Hipotensión ortostática, caídas y síncope y Reacciones Adversas - Otras reacciones adversas observadas durante la evaluación de ensayos clínicos antes y después de la comercialización de duloxetina]. Síndrome Serotoninérgico: Se ha reportado desarrollo de síndrome serotoninérgico que representan un peligro potencial para la vida con el uso de IRSN e ISRS, incluyendo el tratamiento solo con DUXETIN®, pero principalmente con el uso combinado de otros fármacos serotoninérgicos (incluyendo triptanos, antidepresivos tricíclicos, fentanilo, litio, tramadol, triptófano, buspirona, anfetaminas y Hierba de San Juan) y con fármacos que difi cultan el metabolismo de la serotonina (particularmente IMAO, ambos aquellos que intentan tratar desórdenes psiquiátricos y también otros como la linezolida y el azul de metileno intravenoso). Los síntomas del síndrome serotoninérgico pueden incluir cambios en el estado mental (p.ej., agitación, alucinaciones, delirium y coma), inestabilidad autónoma (p.ej., taquicardia, tensión arterial lábil, mareos, diaforesis, enrojecimiento, hipertermia), síntomas neuromusculares (p. ej., temblores, rigidez, mioclonus, hiperrefl exia, descoordinación), convulsiones y/o síntomas gastrointestinales (p.ej., náusea, vómitos, diarrea). Los pacientes deben ser monitoreados para detectar la aparición del síndrome serotoninérgico. El uso concomitante de DUXETIN® con IMAOs que intentan tratar desórdenes psiquiátricos está contraindicado. DUXETIN® tampoco debe ser iniciado en pacientes que están siendo tratados con IMAOs tales como linezolida o azul de metileno intravenoso. Todos los reportes con azul de metileno que proporcionan información sobre la vía de administración incluyen administración intravenosa en un rango de dosis que va de 1mg/kg a 8 mg/kg. No hay reportes que indiquen administración de azul de metileno por otras vías (tal como tabletas orales o inyección tisular local) o a dosis menores. Podría haber circunstancias donde sea necesario iniciar un tratamiento con un IMAO tal como linezolida o azul de metileno intravenoso en un paciente que está tomando DUXETIN®. DUXETIN® debe ser discontinuado antes de iniciar el tratamiento con el IMAO [ver Dosifi cación y Modo de Administración y Contraindicaciones]. Si el uso concomitante de DUXETIN® con otras drogas seronotinérgicas incluyendo triptanos, antidepresivos tricíclicos, fentanilo, litio, tramadol, buspirona, triptófano, anfetaminas y Hierba de San Juan está clínicamente garantizado, entonces los pacientes deben ser advertidos del potencial riesgo incrementado para síndrome serotoninérgico, especialmente durante el inicio del tratamiento y cuando se aumenten las dosis [véase Interacciones]. El tratamiento con DUXETIN® y cualquier agente serotoninérgico debe ser discontinuado de inmediato en caso ocurriesen los eventos antes mencionados e iniciar un tratamiento sintomático de soporte. Sangrado anormal: Los ISRS e IRSN, incluyendo DUXETIN®, pueden elevar el riesgo de episodios de sangrado. El uso concomitante del ácido acetil salicílico, fármacos antiinfl amatorios no esteroides, warfarina y otros anticoagulantes pueden sumarse a este riesgo. Los reportes de casos y los estudios epidemiológicos (caso-control y diseño de cohorte) han demostrado una asociación entre el uso de fármacos que interfi eren con la recaptación de la serotonina y la ocurrencia de hemorragias gastrointestinales. Los episodios de hemorragia relacionados al uso de ISRS e IRSN van desde equimosis, hematomas, epistaxis y petequias hasta hemorragias potencialmente fatales. Se debe advertir a los pacientes sobre el riesgo de hemorragias asociadas al uso concomitante de DUXETIN® y AINE, ácido acetil salicílico, u otros fármacos que afectan la coagulación. Reacciones dermatológicas severas: Durante el tratamiento con DUXETIN® pueden producirse reacciones dermatológicas severas, incluyendo el eritema multiforme y el síndrome Stevens-Johnson (SJS por sus siglas en inglés). El índice de reporte de SJS asociado al uso de DUXETIN® excede la tasa de incidencia en los antecedentes de la población general para los casos de esta seria reacción dermatológica (1 a 2 casos por cada millón de personas por año). Por lo general se acepta que el índice de reporte esté subestimado debido a la baja incidencia de reportes. Se debe discontinuar el uso de DUXETIN® ante la primera aparición de ampollas, erupciones cutáneas con descamación, erosiones mucosas, o cualquier otra señal de hipersensibilidad en caso no se haya identifi cado otra etiología. Discontinuación del tratamiento con DUXETIN®: Los síntomas que causan la discontinuación se han evaluado de manera sistemática en pacientes que toman DUXETIN®. Luego de su discontinuación abrupta o reducción gradual en adultos en ensayos clínicos controlados por placebo, ocurrieron los siguientes síntomas en un 1% de pacientes o más y a un índice signifi cativamente mayor en pacientes tratados con DUXETIN® en comparación con los que fueron discontinuados del placebo: mareos, náusea, cefalea, parestesia, fatiga, vómito, irritabilidad, insomnio, diarrea, ansiedad e hiperhidrosis. Durante la comercialización de otros ISRS e IRSN (inhibidores de la recaptación de serotonina y norepinefrina), han habido reportes espontáneos de eventos adversos que ocurren luego de la discontinuación de estos fármacos, especialmente cuando es abrupta, incluyendo los siguientes: estado de ánimo disfórico, irritabilidad, agitación, mareo, disturbios sensoriales (p.ej., parestesias tales como sensación de shocks eléctricos), ansiedad, confusión, cefalea, letargo, labilidad emocional, insomnio, hipomanía, tinnitus, y convulsiones. Aunque estos eventos son normalmente espontáneos, algunos han sido reportados como severos. Los pacientes deben ser monitoreados ante estos síntomas al discontinuar el tratamiento con DUXETIN®. Se recomienda una reducción gradual de la dosis en lugar de una suspensión abrupta siempre que sea posible. Si ocurriesen síntomas intolerables luego de reducir la dosis o al discontinuar el tratamiento, entonces se puede considerar reanudar la dosis previamente prescrita. Posteriormente, el médico puede continuar reduciendo la dosis pero a un índice más gradual [véase Dosifi cación y Modo de Administración]. Activación de manía/hipomanía: En los ensayos controlados por placebo en adultos entre pacientes con trastorno depresivo mayor, se reportó la activación de casos de manía o hipomanía en un 0.1% (4/3779) de pacientes tratados con DUXETIN® y un 0.04% (1/2536) de pacientes tratados con placebo. No se reportó ninguna activación de manía o hipomanía en casos de ensayos controlados por placebo para pacientes con Dolor Neuropático Periférico de origen Diabético, Trastorno de Ansiedad Generalizada, Fibromialgia, o Dolor Musculoesquelético Crónico. En una pequeña proporción de pacientes con trastornos del estado de ánimo, tratados con otros fármacos comercializados efectivos para el tratamiento del trastorno depresivo mayor, se reportó la activación de casos de manía o hipomanía. Como con estos otros agentes, DUXETIN® debe ser usado cuidadosamente en pacientes con un historial de manía. Convulsiones: DUXETIN® no se ha evaluado de manera sistemática en pacientes con un trastorno de convulsiones, y dichos pacientes han sido excluidos de los estudios clínicos. En ensayos clínicos controlados por placebo en adultos, las convulsiones ocurrieron en un 0,02% (3/12.722) de pacientes tratados con DUXETIN® y un 0,01% (1/9513) de pacientes tratados con placebo. DUXETIN® debe ser prescrito con cuidado en pacientes con un historial de trastorno de convulsiones. Efectos sobre la tensión arterial: En ensayos clínicos en adultos controlados por placebo por indicaciones desde el inicio hasta el fi nal, el tratamiento con DUXETIN® estuvo asociado a aumentos promedio de 0.5 mm Hg para la tensión arterial sistólica y 0.8 mm Hg para la tensión arterial diastólica en comparación con la reducción promedio de 0.6 mm Hg en la sistólica y 0.3 mm Hg en la diastólica en pacientes tratados con placebo. No hubo ninguna diferencia signifi cativa en la frecuencia de la tensión arterial alta sostenida (3 visitas consecutivas). En un estudio clínico farmacológico diseñado para evaluar los efectos de DUXETIN® sobre varios parámetros, incluyendo la presión sanguínea en dosis supraterapéuticas con una titulación acelerada de la dosis, hubo evidencia de aumentos en la tensión arterial supina en dosis superiores a 200 mg dos veces al día. Con la dosis diaria más alta de 200 mg dos veces al día, el aumento en el índice promedio del pulso fue de 5.0 a 6.8 latidos y los aumentos de la tensión arterial promedio fueron de 4.7 a 6.8 mm Hg (sístole) y 4.5 a 7 mm Hg (diástole) hasta 12 horas después de administrada la dosis. Se debe tomar la tensión arterial antes de iniciar el tratamiento y de forma periódica durante el mismo [véase Reacciones Adversas]. Interacciones medicamentosas clínicamente importantes: Tanto el CYP1A2 como el CYP2D6 son responsables del metabolismo de DUXETIN®. Potencial de que DUXETIN® se vea afectada por otros fármacos: Inhibidores del CYP1A2. Se debe evitar la coadministración de DUXETIN® con potentes inhibidores del CYP1A2 [véase Interacciones]. Inhibidores del CYP2D6. Ya que el CYP2D6 está involucrado en el metabolismo de la duloxetina, se espera que el uso concomitante de DUXETIN® con potentes inhibidores del CYP2D6 dé como resultado, y así sucede, mayores concentraciones (en un promedio de 60%) de DUXETIN® [véase Interacciones]. Potencial de DUXETIN® para afectar a otros fármacos: Fármacos metabolizados por CYP2D6. Se debe tener sumo cuidado al coadministrar DUXETIN® con fármacos que son altamente metabolizados por CYP2D6 y que tienen un índice terapéutico limitado, incluyendo ciertos antidepresivos (antidepresivos tricíclicos [ATC], tales como la nortriptilina, la amitriptilina y la imipramina), fenotiazinas y antirrítmicos del tipo 1C (p.ej., propafenona fl ecainida). Es posible que se deban monitorear las concentraciones de plasma de ATC y la dosis de los ATC puede necesitar reducirse si un ATC es coadministrado con DUXETIN®. Debido al riesgo de serias arritmias ventriculares y muerte súbita potencialmente asociadas a los altos niveles de plasma en la tioridazina, DUXETIN® y tioridazina no deben ser administrados en combinación [véase Interacciones]. Otras interacciones medicamentosas clínicamente importantes: Alcohol. El uso concomitante de DUXETIN® con el consumo constante de alcohol puede estar asociado a un daño hepático severo. Por esta razón, DUXETIN® no debe ser prescrito a pacientes que consuman alcohol de manera considerable [véase Advertencias y Precauciones Especiales de Uso e Interacciones]. Fármacos que actúan sobre el SNC. Considerando los efectos primarios de DUXETIN® sobre el SNC, éste debe ser usado con cautela cuando es combinado con otros fármacos en el tratamiento del sistema nervioso central, incluyendo aquéllos con un mecanismo de acción similar [véase Advertencias y Precauciones Especiales de Uso e Interacciones]. Hiponatremia: La hiponatremia puede ocurrir como resultado del tratamiento con ISRS e IRSN, incluyendo DUXETIN®. En muchos casos, esta hiponatremia aparece como el resultado del síndrome de secreción inadecuada de la hormona antidiurética (SSIHAD). Se han reportado casos de sodio en el suero a niveles menores de 110 mmol/L que aparentemente eran reversibles al discontinuar DUXETIN®. Los pacientes ancianos pueden encontrarse en un mayor riesgo de desarrollar hiponatremia con ISRS e IRSN. Asimismo, los pacientes que toman diuréticos o que por otras razones poseen volúmenes menores pueden encontrarse en un mayor riesgo [véase Advertencias y Precauciones Especiales de Uso - Uso en poblaciones específi cas]. La discontinuación de DUXETIN® se debe considerar en pacientes con hiponatremia sintomática y se debe implementar además una apropiada intervención médica. Los signos y síntomas de hiponatremia incluyen cefalea, difi cultad para concentrarse, difi cultad en la memoria, confusión, debilidad, e inestabilidad, los cuales pueden producir caídas. Casos más severos y/o agudos se han asociado a alucinaciones, síncope, convulsiones, coma, paros respiratorios, y muerte. Uso en pacientes con una enfermedad concomitante: La experiencia clínica de DUXETIN® en pacientes con una enfermedad concomitante sistémica es limitada. No existe información sobre el efecto que las alteraciones en la motilidad gástrica pueden tener sobre la estabilidad del recubrimiento entérico de DUXETIN®. En condiciones extremadamente ácidas, DUXETIN®, sin la protección de su recubrimiento entérico, puede sufrir una hidrólisis y formar naftol. Se debe tener mucho cuidado al usar DUXETIN® en pacientes con condiciones que puedan retrasar el vaciado gástrico (p.ej., algunos diabéticos). DUXETIN® no ha sido evaluado de manera sistemática en pacientes con un historial reciente de infarto del miocardio o una enfermedad ine stable de la arteria coronaria. Los pacientes con estos diagnósticos normalmente fueron excluidos de los estudios clínicos durante las pruebas de pre comercialización del producto. Insufi ciencia Hepática: DUXETIN® no debería ser usado ordinariamente en pacientes con insufi ciencia hepática (ver Dosifi cación y Modo de Administración, Advertencias y Precauciones, Uso en poblaciones especifi cas). Insufi ciencia renal severa: DUXETIN® no de
bería ser usado ordinariamente en pacientes con una enfermedad renal o insufi ciencia renal severa en su etapa fi nal (depuración de creatinina < 30 mL/min). En pacientes con una enfermedad renal en su etapa fi nal (requieren diálisis) se observa un incremento de las concentraciones plasmáticas de la duloxetina, especialmente de sus metabolitos (ver Dosifi cación y Uso en poblaciones especifi cas). Glaucoma controlado de ángulo estrecho: En ensayos clínicos, DUXETIN® estuvo asociado a un mayor riesgo de midriasis; por ello, debe ser usado con cuidado en pacientes con glaucoma controlado de ángulo estrecho (ver Contraindicaciones). Control glucémico de pacientes con diabetes: Tal como se observó en ensayos Dolor Neuropático Periférico de origen Diabético, el tratamiento con DUXETIN® perjudica el control glucémico de algunos pacientes diabéticos. En tres ensayos clínicos de DUXETIN® para el tratamiento del dolor neuropático asociado a la neuropatía diabética periférica, la duración promedio de la diabetes fue de aproximadamente 12 años, el nivel promedio de glucosa en la sangre en ayunas en un inicio era de 176 mg/dL, y la hemoglobina A1c (HbA1c) promedio en un inicio era 7,8%. En la semana 12 de la fase del tratamiento agudo de estos estudios, DUXETIN® estuvo asociado a un ligero incremento en el nivel promedio de la glucosa en la sangre en ayunas en comparación con el placebo. En la fase de extensión de estos estudios, la cual duró 52 semanas, el nivel promedio de glucosa en la sangre en ayunas aumentó en 12 mg/dL en el grupo que tomó DUXETIN® y disminuyó 11,5 mg/dL en el grupo de atención de rutina. La HbA1c aumentó en 0,5% en el grupo DUXETIN® y 0,2% en los grupos de atención de rutina. Retención y Vacilación urinaria: DUXETIN® pertenece a una clase de fármacos de los cuales se sabe que afectan la resistencia uretral. Si se desarrollan síntomas de vacilación urinaria durante el tratamiento con DUXETIN®, debe considerarse la posibilidad de que puedan estar relacionados al fármaco. En la experiencia post comercial, se observaron casos de retención urinaria. En algunos casos de retención urinaria asociada al uso de DUXETIN®, se necesitó hospitalizar al paciente o realizar un cateterismo. Pruebas de laboratorio: No se recomienda ninguna prueba específi ca de laboratorio. Uso en Poblaciones Específicas: Embarazo: Resumen de Riesgos - No hay estudios adecuados y bien controlados de la administración de DUXETIN® en mujeres embarazadas. En estudios en animales con duloxetina, los pesos fetales disminuyeron, pero no hubo teratogenicidad en ratas y conejas preñadas a dosis orales administradas hasta 4 y 7 veces la dosis máxima recomendada en humanos (DMRH) de 120 mg/día, respectivamente durante el periodo de organogénisis. Cuando se administró duloxetina por vía oral a una dosis de 2 veces la DMRH a ratas embarazadas durante la gestación y lactancia, se disminuyeron el peso al nacer y la supervivencia de 1 día postnatal de las crías. A esta dosis se observó comportamientos compatibles con mayor reactividad, como el aumento de la respuesta de sobresalto al ruido y la disminución de la habituación a la actividad locomotora. No se afectó de manera adversa el crecimiento post-destete. DUXETIN® se debe utilizar durante el embarazo sólo si el benefi cio potencial justifi ca el riesgo potencial para el feto. Consideraciones Clínicas: Reacción Adversa Fetal/Neonatal - Recién nacidos expuestos durante el embarazo a la serotonina - inhibidores selectivos de la recaptación de norepinefrina (ISRN) o inhibidores selectivos de la recaptación de serotonina (ISRS) han desarrollado complicaciones que requieren hospitalización prolongada, asistencia respiratoria, y alimentación por sonda, que pueden surgir inmediatamente tras el nacimiento. Los hallazgos clínicos reportados han incluido problemas respiratorios, cianosis, apnea, convulsiones, inestabilidad en la temperatura, difi cultad para comer, vómitos, hipoglicemia, hipotonía, hipertonía, hiperrefl exia, temblores, nerviosismo, irritabilidad, y llanto constante. Estas características son consistentes con un efecto tóxico directo de los ISRS o IRNS o, posiblemente, un síndrome de discontinuación del fármaco. Se debe considerar que, en algunas ocasiones, el caso clínico es consistente con el síndrome serotoninérgico [véase Advertencias y Precauciones Especiales de Uso]. Datos: Datos en animales. En estudios de reproducción con animales, se ha demostrado que la duloxetina tiene efectos adversos sobre el embrión/feto y el desarrollo postnatal. Cuando de manera oral se administró duloxetina a ratas y conejas preñadas durante el período de organogénesis, no hubo evidencia de teratogenicidad con dosis de hasta 45 mg/kg/día (4 veces la dosis máxima recomendada en humanos (DMRH) de 120 mg/día sobre una base de mg/m2, en ratas; 7 veces la DMRH en conejos hembra). No obstante, los pesos fetales disminuyeron con esta dosis, con una dosis sin efecto de 10 mg/kg/día aproximadamente igual a la DMRH en ratas; 2 veces la DMRH en conejos hembra. Cuando se administró duloxetina de manera oral a ratas preñadas durante el período de gestación y lactancia, la sobrevivencia de las crías en la etapa post parto a 1 día y el peso corporal de las crías al momento del nacimiento y durante la lactancia disminuyeron con una dosis de 30 mg/kg/día (2 veces la DMRH); la dosis sin efecto fue de 10 mg/kg/día. Además, se observaron conductas consistentes con el aumento de la reactividad, tales como más respuestas de manera sobresaltada a ruidos y pérdida del hábito a las actividades locomotrices, en crías después de la exposición materna con 30 mg/kg/día. El crecimiento posterior al destete y el rendimiento reproductivo de la progenie no se vieron afectados de manera adversa por el tratamiento con duloxetina de las madres. Madres lactantes: Resumen de Riesgos: DUXETIN® está presente en la leche humana. En un estudio publicado, se administró DUXETIN® a mujeres lactantes que estaban destetando a sus infantes. En el estado de equilibrio, la concentración de DUXETIN® en la leche materna fue de aproximadamente 25% de la del plasma materno. La dosis diaria estimada de un infante fue de aproximadamente 0,14% de la dosis materna. Los benefi cios de la alimentación con leche materna para la salud y el desarrollo deben considerarse junto con la necesidad clínica de DUXETIN® para la madre y cualquier efecto adverso potencial del medicamento o de la condición materna subyacente sobre el infante. Tener precaución al administrar DUXETIN® a una mujer lactante. Datos: La disposición de DUXETIN® fue estudiada en 6 madres lactantes que tenían por lo menos 12 semanas de post parto y que habían escogido a destetar a sus infantes. Se les administró 40 mg de DUXETIN® dos veces al día durante 3.5 días. A una media de 3 horas después de la dosis se produjo la concentración pico medida en la leche materna. La cantidad de DUXETIN® en la leche materna fue de aproximadamente 7mcg/día con esta dosis; la dosis diaria estimada para el infante fue de aproximadamente 2 mcg/Kg/día. No se examinó la presencia de metabolitos de DUXETIN® en la leche materna. Ya que aún se desconoce la seguridad de duloxetina en infantes, no se recomienda dar de lactar mientras se toma DUXETIN®. Uso pediátrico: No se debe utilizar en niños y adolescentes menores de 18 años. Trastorno Depresivo Mayor: No fue demostrada efi cacia en 2 estudios controlados por placebo de 10 semanas de duración en 800 pacientes pediátricos con Trastorno Depresivo Mayor con edades comprendidas entre 7 a 17 años. Ni DUXETIN® ni un control activo (indicado para el tratamiento de depresión en pacientes pediátricos), fue superior al placebo. Por lo tanto, no se ha establecido la seguridad y efi cacia de DUXETIN® en pacientes pediátricos menores de 18 años con trastorno depresivo mayor. Las reacciones adversas observadas con mayor frecuencia en los estudios clínicos incluyeron náusea, dolor de cabeza, disminución de peso y dolor abdominal. Se debe realizar monitoreo regular de peso y talla en niños y adolescentes tratados con IRSN tal como DUXETIN® [véase Reacciones Adversas]. Datos en Animales: La administración de duloxetina a ratas jóvenes desde el día 21 (destete) hasta el día 90 (adulto) resultó en una disminución del peso corporal que persistió hasta su adultez, pero se recuperó cuando el tratamiento fue discontinuado; ligero retraso en la maduración sexual en hembras sin ningún efecto en la fertilidad y retraso en el aprendizaje de tareas complejas en la adultez lo cual no fue observado luego que el tratamiento fue discontinuado. Estos efectos fueron observados a altas dosis de 45 mg/kg/día (2 veces la DMRH para un niño); el nivel de no efecto fue a 20 mg/kg/día (1 vez la DMRH para un niño). Uso geriátrico: De los 2.418 pacientes en estudios clínicos para Trastorno Depresivo Mayor previos a la comercialización de DUXETIN®, un 5,9% (143) tenían 65 años de edad o más. De los 1.041 pacientes en estudios previos a la comercialización para Dolor Crónico en la Zona Lumbar, un 21,2% (221) tenían 65 años de edad o más. De los 487 pacientes en estudios previos a la comercialización para Osteoartritis, un 40,5% (197) tenían 65 años de edad o más. De los 1,074 pacientes en estudios previos a la comercialización para Dolor Neuropático Periférico de origen Diabético, un 33% (357) tenían 65 años de edad o más. De los 1,761 pacientes en estudios previos a la comercialización para Fibromialgia un 7,9% (140) tenían 65 años de edad o más. Diferencias generales en la seguridad o efectividad no fueran comúnmente observadas entre estos sujetos y sujetos más jóvenes en los estudios para Trastorno Depresivo Mayor, Trastorno Ansiedad Generalizada, Dolor Neuropático Periférico de origen Diabético, Fibromialgia, Osteoartritis y Dolor Crónico en la Zona Lumbar, sin embargo, no se puede descartar la mayor sensibilidad de algunos individuos mayores. Se han asociado los ISRS y los IRNS, incluso DUXETIN® con casos de hiponatremia clínicamente signifi cativa en pacientes de edad avanzada, así que pueden tener un mayor riesgo de presentar este evento adverso [véase Advertencias y Precauciones Especiales de Uso]. En un análisis de datos de todos los estudios controlados por placebo, los pacientes tratados con DUXETIN® reportaron una mayor tasa de caídas en comparación con los pacientes tratados con placebo. El aumento del riesgo parece ser proporcional al riesgo subyacente de un paciente de caídas. El riesgo subyacente parece aumentar progresivamente con la edad. Como los pacientes de edad avanzada tienden a tener una mayor prevalencia de factores de riesgos de caídas, tales como medicamentos, comorbilidad médica y trastornos de la marcha, el impacto del aumento de la edad por si solo para las caídas durante el tratamiento con DUXETIN® no está claro. Se han reportado caídas con consecuencias graves, como fracturas óseas y hospitalizaciones [véase Advertencias y Precauciones Especiales de Uso y Reacciones Adversas]. Se compararon las farmacocinéticas de la duloxetina después de una dosis única de 40 mg en mujeres sanas de edad avanzada (de 65 a 77 años de edad) y en mujeres sanas de mediana edad (de 32 a 50 años de edad). No hubo diferencia en la Cmax, sin embargo, la AUC de la duloxetina fue un poco (cerca de 25%) superior y la vida media fue aproximadamente 4 horas más en mujeres de edad avanzada. Los análisis farmacocinéticos en la población sugieren que los valores típicos para la depuración disminuyen aproximadamente un 1% cada año de edad entre los 25 y 75 años de edad; pero la edad como factor predictivo solo cuenta para un pequeño porcentaje de variabilidad entre un paciente y otro. Los ajustes a las dosis con base en la edad del paciente no son necesarios. Género: La vida media de la duloxetina es similar en hombres y mujeres. No son necesarios los ajustes a las dosis en base al género. Fumadores: La biodisponibilidad (AUC) de la duloxetina parece reducirse en aproximadamente un tercio en fumadores. No se recomiendan las modifi caciones de las dosis en el caso de fumadores. Raza: No se ha realizado un estudio farmacocinético específi co para investigar variaciones en distintas etnias. Insufi ciencia hepática: Los pacientes con insufi ciencia hepática clínicamente evidente han disminuido el metabolismo de la duloxetina y la eliminación. Después de una sola dosis de 20 mg de DUXETIN®, 6 pacientes cirróticos con una insufi ciencia hepática moderada (escala de Child-Pugh, clases B) tuvieron una depuración promedio de la duloxetina plasmática, de aproximadamente un 15% de la correspondiente a sujetos sanos de la misma edad y género, con un aumento de 5 veces la exposición promedio (AUC). Aunque los niveles de Cmax fueron similares en los pacientes cirróticos a los normales, la vida media fue más o menos 3 veces más extensa [véase Dosifi cación y Modo de Administración y Advertencias y Precauciones Especiales de Uso]. Insuficiencia renal severa: Existe información limitada sobre los efectos de la duloxetina en pacientes con defi ciencia renal en su etapa fi nal (ESRD). Después de una sola dosis de duloxetina, los valores de Cmax y AUC fueron aproximadamente 100% mayores en pacientes con una enfermedad renal en su etapa fi nal que reciben hemodiálisis intermitente crónica que en pacientes con funciones renales normales. La vida media en eliminación, sin embargo, fue similar en ambos grupos. Las AUC de los mayores metabolitos circulantes, 4-hidroxi duloxetina glucuronida y 5-hidroxi, 6-sulfato de duloxetina metoxia, que normalmente se excretan en la orina, fueron de aproximadamente 7-9 veces mayor y se espera que aumenten más con dosis múltiples. Los análisis farmacocinéticos en la población sugieren que los grados leves a moderados de insufi ciencia renal (ClCr 30-80 mL/min estimado) no tienen un efecto signifi cativo sobre la depuración aparente de la duloxetina [véase Dosifi cación y Modo de Administración].
Dependencia.
Abuso: En estudios con animales, la duloxetina no demostró tener el potencial de abuso tipo barbitúrico (depresivo). Si bien DUXETIN® no ha sido estudiado de manera sistemática en humanos por su potencial de abuso, nada hace indicar que hubiese una conducta de búsqueda de fármacos en los ensayos clínicos. Sin embargo, no es posible predecir con base en la experiencia de la pre-comercialización el grado de mal uso, desvío, y/o abuso que se le puede dar a un fármaco activo en el Sistema Nervioso Central una vez que sea comercializado. Consecuentemente, los médicos deben evaluar muy cuidadosamente a los pacientes ante un historial de abuso de fármacos y seguir a esos pacientes de cerca, observarlos ante señales de mal uso o abuso de DUXETIN® (p.ej., desarrollo de tolerancia, aumento de la dosis, conducta de búsqueda del fármaco). Dependencia: En estudios de farmacodependencia, la duloxetina no demostró tener el potencial de producir dependencia en ratas.
Interacciones.
Tanto el CYP1A2 como el CYP2D6 son responsables del metabolismo de la duloxetina. Inhibidores del CYP1A2: Cuando duloxetina de 60 mg fue coadministrada con fl uvoxamina de 100 mg, un inhibidor potente del CYP1A2, a sujetos varones (n=14) el AUC de la duloxetina aumentó aproximadamente 6 veces, los valores de Cmáx aumentaron alrededor de 2.5 veces, y la duloxetina t1/2 incrementó aproximadamente 3 veces. Otros fármacos que inhiben el metabolismo del CYP1A2 incluyen cimetidina y antimicrobianos quinolónicos tales como ciprofl oxacina y enoxacina [véase Advertencias y Precauciones Especiales de Uso]. Inhibidores del CYP2D6: El uso concomitante de duloxetina (40 mg una vez al día) con paroxetina (20 mg una vez al día) elevó la concentración del AUC de la duloxetina en aproximadamente 60%, y se esperan mayores niveles de inhibición con dosis más altas de paroxetina. Se deben esperar efectos similares con otros inhibidores potentes del CYP2D6 (p.ej., fl uoxetina, quinidina) [véase Advertencias y Precauciones Especiales de Uso]. Inhibición dual del CYP1A2 y el CYP2D6: La administración concomitante de duloxetina 40 mg dos veces al día con fl uvoxamina 100 mg, un potente inhibidor del CYP1A2, a sujetos con metabolizadores pobres del CYP2D6 (n=14) produjo un aumento de 6 veces del AUC de la duloxetina y Cmáx. Fármacos que interfi eren con la hemostasia (p.ej., AINE, ácido acetil salicílico y warfarina): La liberación de serotonina por las plaquetas juega un importante papel en la hemostasia. Estudios epidemiológicos del caso control y del diseño de la cohorte que demostraron una asociación entre el uso de fármacos psicotrópicos que interfi eren con la recaptación de serotonina y la ocurrencia de hemorragia gastrointestinal superior también han demostrado que el uso simultáneo de AINE o ácido acetil salicílico puede potenciar el riesgo de hemorragias. Se han reportado efectos alterados de los anticoagulantes, incluyendo una mayor hemorragia, al administrar ISRS e IRSN junto con warfarina. La administración concomitante de warfarina (2-9 mg una vez al día) bajo condiciones de estado estable con duloxetina de 60 o 120 mg una vez al día por un período de hasta 14 días en sujetos sanos (n=15) no cambió de manera signifi cativa el INR desde el inicio (los cambios promedio de INR variaron entre 0.05 a +0.07). La farmacocinética (AUCt,ss, Cmax,ss, o tmax,ss) total de la warfarina (golpe proteico más fármaco libre) para la warfarina R- y S- no fue alterada por la duloxetina. Debido al efecto potencial de la duloxetina sobre las plaquetas, los pacientes que reciben una terapia de warfarina deben ser observados con cuidado al iniciar o discontinuar la duloxetina [véase Advertencias y Precauciones Especiales de Uso]. Lorazepám: Bajo condiciones de estado estable para la duloxetina (60 mg c/12 horas) y lorazepám (2 mg c/12 horas), la farmacocinética de la duloxetina no se vio afectada por la coadministración. Temazepám. Bajo condiciones de estado estable para la duloxetina (20 mg cada noche) y temazepám (30 mg cada noche), la farmacocinética de la duloxetina no se vio afectada por la coadministración. Fármacos que afectan la acidez gástrica: DUXETIN® tiene un recubrimiento entérico resistible a la disolución hasta que llega a un segmento del tracto gastrointestinal donde el pH excede 5.5. Bajo condiciones extremadamente ácidas, DUXETIN®, sin la protección del recubrimiento entérico, puede sufrir una hidrólisis y formar naftol. Se debe tener cuidado al usar DUXETIN® en pacientes con condiciones que puedan retrasar el vaciado gástrico (p.ej., algunos diabéticos). Los fármacos que elevan el pH gastrointestinal pueden causar una liberación prematura de la duloxetina. No obstante, la coadministración de DUXETIN® con antiácidos que contienen aluminio y magnesio (51 mEq) o DUXETIN® con famotidina, no tuvo un efecto signifi cativo sobre el índice o el grado de absorción de la duloxetina después de administrar una dosis oral de 40 mg. Se desconoce si la administración concomitante de inhibidores elevadores del protón afecta la absorción de la duloxetina [véase Advertencias y Precauciones Especiales de Uso]. Fármacos metabolizados por CYP1A2: Los estudios de interacción de fármacos in vitro demuestran que la duloxetina no induce la actividad del CYP1A2. Por consiguiente, no se anticipa un aumento en el metabolismo de los sustratos de CYP1A2 (p.ej., teofi lina, cafeína) como resultado de una inducción, aunque no se han realizado estudios clínicos sobre la inducción. La duloxetina es un inhibidor de la isoforma CYP1A2 en estudios in vitro, y en dos estudios clínicos el aumento promedio (90% de intervalo de confi anza) en el AUC de la teofilina fue de 7% (1% - 15%) y 20% (13% - 27%) cuando se coadministró con duloxetina (60 mg dos veces al día). Fármacos metabolizados por CYP2D6: Duloxetina es un inhibidor moderado del CYP2D6. Cuando la duloxetina fue administrada (en una dosis de 60 mg dos veces al día) junto con una sola dosis de 50 mg de desipramina, un sustrato del CYP2D6, el AUC de la desipramina aumentó 3 veces [véase Advertencias y Precauciones Especiales de Uso]. Fármacos metabolizados por CYP2C9: Los resultados de los estudios in vitro demuestran que la duloxetina no inhibe la actividad. En un estudio clínico, la farmacocinética de la S-warfarina, un sustrato del CYP2C9, no se vio afectada de manera signifi cativa por la duloxetina [véase Interacciones]. Fármacos metabolizados por CYP3A: Los resultados de los estudios in vitro demuestran que la duloxetina no inhibe o induce la actividad del CYP3A. Por consiguiente, no se anticipa un aumento o disminución del metabolismo de los sustratos de CYP3A (p.ej., anticonceptivos orales y otros agentes esteroides) como resultado de una inducción o inhibición, aunque no se han realizado estudios clínicos al respecto. Fármacos metabolizados por CYP2C19: Los resultados de estudios in vitro demuestran que la duloxetina no inhibe la actividad del CYP2C19 en concentraciones terapéuticas. La inhibición del metabolismo de sustratos de CYP2C19 por consiguiente no puede anticiparse, aunque no se han realizado estudios clínicos al respecto. Inhibidores de la monoaminoxidasa (IMAOs): [Véase Dosifi cación y Modo de Administración, Contraindicaciones y Advertencias y Precauciones Especiales de Uso]. Fármacos serotoninérgicos: [Véase Dosificación y Modo de Administración, Contraindicaciones y Advertencias y Precauciones Especiales de Uso]. Alcohol: Cuando se administró DUXETIN® y etanol en un lapso de varias horas entre sí para que las concentraciones pico de cada uno coincida, DUXETIN® no agudizó las discapacidades mentales y motrices causadas por el alcohol. En la base de datos de los ensayos clínicos de DUXETIN®, tres pacientes tratados con DUXETIN® sufrieron un daño hepático manifestado por elevaciones de ALT y la bilirrubina total, con evidencia de obstrucción. El uso intercurrente sustancial de etanol estuvo presente en cada uno de estos casos, y esto pudo haber contribuido a las anomalías observadas [véase Advertencias y Precauciones Especiales de Uso]. Fármacos que actúan a nivel del Sistema Nervioso Central: [véase Advertencias y Precauciones Especiales de Uso]. Fármacos fuertemente unidos a la proteína plasmática: Puesto que la duloxetina está fuertemente unida a la proteína plasmática, la administración de DUXETIN® a un paciente que está tomando otro fármaco altamente vinculado a la proteína puede causar el incremento de las concentraciones libres del otro fármaco, lo que potencialmente puede producir reacciones adversas. Sin embargo, la coadministración de duloxetina (60 o 120 mg) con warfarina (2-9 mg), un fármaco altamente vinculado a la proteína, no produjo cambios signifi cativos en el INR y en la farmacocinética de la S- o R-warfarina total (vinculada a la proteína más fármaco libre) [véase Interacciones].
Conservación.
Consérvese en lugar fresco y seco a temperatura ambiente inferior a 30°C.
Sobredosificación.
Señales y síntomas: En la experiencia posterior a la comercialización, se han reportado resultados fatales debido a agudas sobredosis, principalmente por la combinación de sobredosis, pero también con duloxetina sola, en dosis tan pequeñas de 1000mg. Las señales y síntomas de sobredosis (duloxetina sola o con fármacos combinados) incluyeron somnolencia, coma, síndrome serotoninérgico, convulsiones, síncope, taquicardia, hipotensión, hipertensión y vómitos. Manejo de la sobredosis: No existe un antídoto específi co para DUXETIN®, pero si el síndrome serotoninérgico persiste, se puede considerar un tratamiento específi co (p.ej., con ciproheptadina y/o control de la temperatura). En el caso de una sobredosis aguda, el tratamiento debe consistir en las medidas generales empleadas en el manejo de una sobredosis con cualquier otro fármaco. Se debe asegurar una adecuada respiración, oxigenación y ventilación, y se deben monitorear el ritmo cardíaco y los signos vitales. No se recomienda la inducción del vómito. Se puede indicar un lavado gástrico mediante una sonda orogástrica con la adecuada protección de las vías respiratorias si se practica poco tiempo después de la ingesta o en pacientes sintomáticos. El carbón activado puede servir para limitar la absorción de la duloxetina del tracto gastrointestinal. La administración de carbón activado ha demostrado disminuir el AUC y el nivel Cmax en un tercio en promedio, aunque algunos sujetos experimentaron un efecto limitado del carbón activado. Debido al gran volumen de distribución de este fármaco, es poco probable que la diuresis forzada, la diálisis, la hemoperfusión y la transfusión sean beneficiosas en estos casos. Al tratar una sobredosis, se debe considerar la posibilidad de la participación de múltiples fármacos. Debe haber sumo cuidado cuando se trata de pacientes que están tomando o recientemente han tomado DUXETIN® y pueden consumir cantidades excesivas de un ATC. En tal caso, una menor depuración del tricíclico parental y/o sus metabolitos activos pueden aumentar la posibilidad de una secuela clínicamente signifi cativa y prolongar el tiempo necesario para una observación médica cercana [véase Advertencias y Precauciones Especiales de Uso e Interacciones]. Ante la eventualidad de una sobredosifi cación, concurrir al hospital más cercano o comunicarse con los Centros de Toxicología: Hospital de Pediatría Dr. Ricardo Gutiérrez: (011) 4962-6666/2247. Hospital Dr. Alejandro Posadas: (011) 4658-7777/4654-6648. Optativamente otros Centros de Intoxicaciones.
Presentación.
DUXETIN® 30: se presenta en envases conteniendo 14 cápsulas con microgránulos con recubrimiento entérico. DUXETIN® 60: se presenta en envases conteniendo 14 y 28 cápsulas con microgránulos con recubrimiento entérico.
Revisión.
04/2018.
Laboratorio que comercializa Duxetin
GADOR →