CORACIL®
GADOR
Hipolipemiante.
Composición.
Cada comprimido de CORACIL® contiene: Ezetimibe 10 mg. Excipientes: Almidón de maíz, Lactosa monohidrato, Povidona, Laurilsulfato de sodio, Croscaramelosa sódica, Estearato de magnesio c.s. Este medicamento contiene lactosa.
Farmacología.
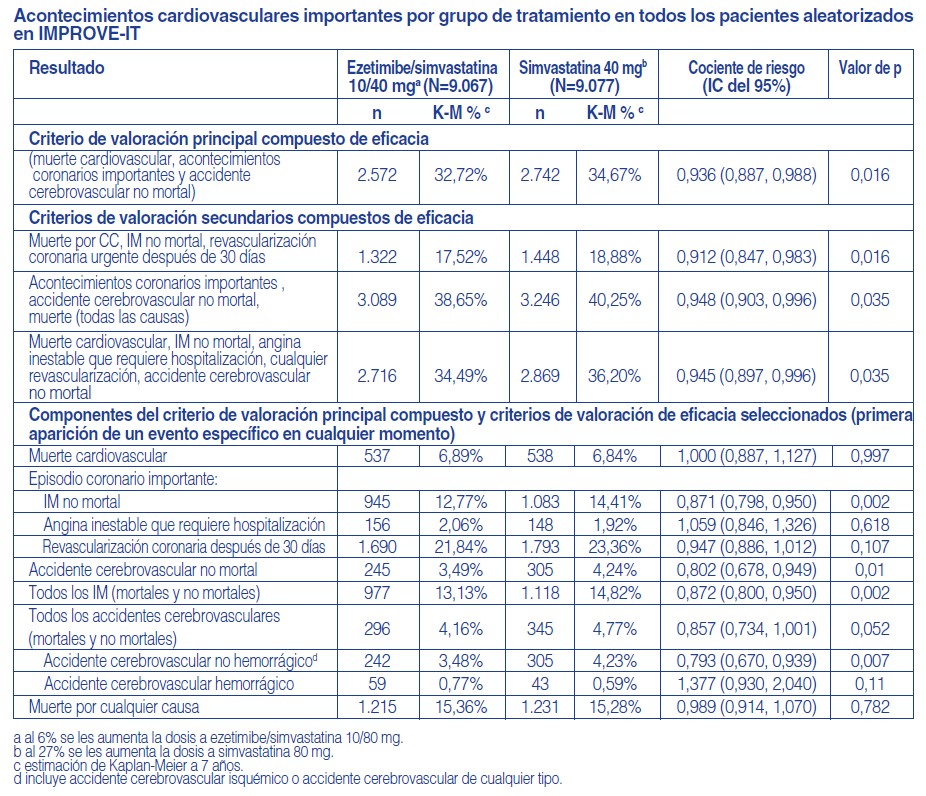
Descripción: CORACIL® (ezetimibe) pertenece a los compuestos hipolipemiantes, que inhiben selectivamente la absorción intestinal de colesterol y fitosteroles relacionados. Ezetimibe disminuye el colesterol sanguíneo por inhibición de la absorción intestinal del colesterol en las vellosidades intestinales donde se concentra. Esta inhibición ocasiona una disminución de aporte de colesterol intestinal al hígado con la consiguiente disminución de los depósitos de colesterol hepáticos y el aumento de la depuración del colesterol de la sangre. Ezetimibe no inhibe la síntesis de colesterol en el hígado, ni aumenta la excreción de ácidos biliares; este mecanismo diferente complementa al de los inhibidores de la HMG-CoA reductasa y al del fenofibrato. En los pacientes con hipercolesterolemia Ezetimibe disminuye el C-total, el C-LDL, las apolipoproteínas B y los triglicéridos y aumenta el C-HDL. La administración concomitante de Ezetimibe con una estatina se complementa y mejora los resultados obtenidos con cualquiera de las dos drogas por separado. Todavía no se ha establecido si Ezetimibe solo o asociado a las estatinas influye sobre la morbilidad y/o la mortalidad cardiovascular. Ezetimibe no modifica la absorción de los triglicéridos, los ácidos grasos, los ácidos biliares, la progesterona, el etinilestradiol, o las vitaminas liposolubles A, D y E. El contenido de colesterol del hígado tiene principalmente tres orígenes. El hígado puede sintetizar colesterol, absorber colesterol de las lipoproteínas circulantes en la sangre, o tomar colesterol absorbido por el intestino delgado. El colesterol intestinal deriva principalmente del colesterol segregado en la bilis y del colesterol dietario. Ezetimibe tiene un mecanismo de acción que se diferencia de otras clases de compuestos para reducir colesterol (inhibidores de la HMG-CoA reductasa, secuestradores de ácidos biliares (resinas), derivados del ácido fíbrico, y estanoles de plantas). Prevención de acontecimientos cardiovasculares: El estudio IMPROVE-IT (IMProved Reduction of Outcomes: Vytorin Efficacy International Trial) fue un estudio multicéntrico, aleatorizado, doble ciego y con control activo realizado en 18.144 pacientes reclutados en los 10 días siguientes a una hospitalización por un síndrome coronario agudo (SCA; infarto agudo de miocardio [IM] o angina inestable [AI]). Los pacientes tenían un C-LDL de 125 mg/dl (≤ 3,2 mmol/l) en el momento en que presentaron el SCA si no habían estado tomando un tratamiento hipolipemiante o de ≤100 mg/dl (≤2,6 mmol/l) si habían estado recibiendo un tratamiento hipolipemiante. Se aleatorizó a todos los pacientes en una proporción de 1:1 para recibir ezetimibe/simvastatina 10/40 mg (n=9.067) o simvastatina 40 mg (n=9.077) y seguidos durante una mediana de 6,0 años. La edad media de los pacientes era de 63,6 años; el 76% eran varones, el 84% eran de raza blanca y el 27% eran diabéticos. El valor medio de C-LDL en el momento que los pacientes presentaron el acontecimiento para ser admitidos en el estudio era de 80 mg/dl (2,1 mmol/l) en los que recibían tratamiento hipolipemiante (n=6.390) y de 101 mg/dl (2,6 mmol/l) en los que no habían recibido tratamiento hipolipemiante previo (n=11.594). Antes de la hospitalización por el acontecimiento de SCA, el 34% de los pacientes recibían tratamiento con estatinas. Al cabo de un año, el C-LDL medio en los pacientes que seguían en tratamiento era de 53,2 mg/dl (1,4 mmol/l) en el grupo de ezetimibe/simvastatina y de 69,9 mg/dl (1,8 mmol/l) en el grupo de simvastatina en monoterapia. Los valores de lípidos se determinaron generalmente en los pacientes que seguían recibiendo el tratamiento del estudio. El criterio de valoración principal fue una combinación de muerte cardiovascular, acontecimientos coronarios importantes (definidos como IM no mortal, AI documentada que requirió hospitalización o cualquier procedimiento de revascularización coronaria realizado al menos 30 días después de la asignación del tratamiento aleatorizado) y accidente cerebrovascular no mortal. El estudio demostró que el tratamiento con ezetimibe, cuando se añade a simvastatina, proporcionó un beneficio mayor en la reducción del criterio de valoración principal compuesto de muerte cardiovascular, acontecimientos coronarios importantes y accidente cerebrovascular no mortal que el tratamiento con simvastatina sola (reducción del riesgo relativo del 6,4%, p=0,016). El criterio de valoración principal se produjo en 2.572 de los 9.067 pacientes del grupo de ezetimibe/simvastatina (tasa de Kaplan-Meier [KM] a los 7 años del 32,72%) y en 2.742 de los 9.077 pacientes del grupo de simvastatina sola (tasa de KM a los 7 años del 34,67%). Cabe esperar que este beneficio incremental para reducir el riesgo de acontecimientos cardiovasculares sea similar con la administración conjunta de otras estatinas. La tasa de mortalidad total no cambió en este grupo de pacientes de alto riesgo. Se observó un beneficio general en todos los tipos de accidente cerebrovascular; sin embargo hubo un pequeño aumento no significativo de la incidencia de accidente cerebrovascular hemorrágico en el grupo de ezetimibe/simvastatina en comparación con el grupo de simvastatina sola. El riesgo de accidente cerebrovascular hemorrágico con ezetimibe administrado conjuntamente con estatinas más potentes no se ha evaluado en estudios de resultados a largo plazo. Por lo general, el efecto del tratamiento con ezetimibe/simvastatina fue similar a los resultados globales en numerosos subgrupos, definidos en función del sexo, la edad, la raza, antecedentes de diabetes, las concentraciones iniciales de lípidos, el tratamiento previo con estatinas, accidente cerebrovascular previo y la hipertensión.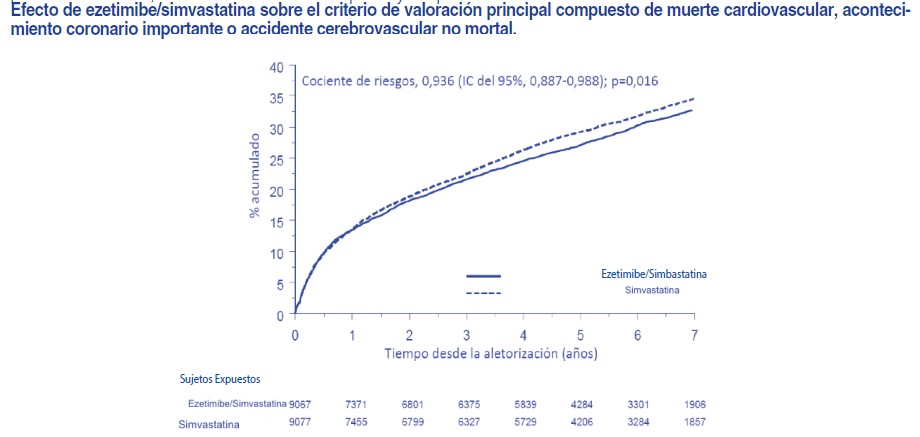
Prevención de acontecimientos vasculares importantes en la enfermedad renal crónica: El Study of Heart and Renal Protection (SHARP), fue un estudio multinacional, aleatorizado, controlado con placebo, doble ciego, realizado en 9.438 pacientes con insuficiencia renal crónica, un tercio de los cuales estaban en diálisis al principio del estudio. Un total de 4.650 pacientes fueron asignados a una combinación a dosis fijas de ezetimibe 10 mg con simvastatina 20 mg y 4.620 a placebo, y seguidos durante una media de 4,9 años. Los pacientes tenían una edad media de 62 años y el 63% eran varones, el 72% era de raza blanca, el 23% eran diabéticos y, en aquellos que no estaban en diálisis, el índice de filtración glomerular medio estimado fue 26,5 ml/min/1,73 m2. No hubo criterio de entrada por lípidos. El C-LDL medio inicial fue de 108 mg/dl. Después de un año, incluyendo a los pacientes que ya no tomaban la medicación del estudio, el C-LDL se redujo en un 26% en relación a placebo por simvastatina 20 mg sola y un 38% con ezetimibe 10 mg en combinación con simvastatina 20 mg. La comparación primaria del protocolo especificado del SHARP fue el análisis por intención de tratar "acontecimientos vasculares importantes" (definido como IM no mortal o muerte cardiaca, accidente cerebrovascular o cualquier procedimiento de revascularización) sólo en aquellos pacientes inicialmente aleatorizados a los grupos de ezetimibe en combinación con simvastatina (n=4.193) o placebo (n=4.191). Los análisis secundarios incluyeron la misma combinación analizada para la cohorte completa aleatorizada (al principio del estudio o en el año 1) para ezetimibe en combinación con simvastatina (n=4.650) o para placebo (n=4.620), así como los componentes de esta combinación. El análisis del criterio principal de valoración demostró que ezetimibe en combinación con simvastatina redujo significativamente el riesgo de acontecimientos vasculares importantes (749 pacientes con acontecimientos en el grupo placebo frente a 639 en el grupo de ezetimibe en combinación con simvastatina) con una reducción del riesgo relativo del 16% (p=0,001). Sin embargo, el diseño de este estudio no tuvo en consideración una contribución separada de la eficacia del componente ezetimibe para reducir significativamente el riesgo de acontecimientos vasculares importantes en pacientes con insuficiencia renal crónica. Ezetimibe en combinación con simvastatina redujo significativamente el riesgo de accidente cerebrovascular y de cualquier revascularización, con diferencias numéricas no significativas a favor de ezetimibe en combinación con simvastatina en infarto de miocardio no mortal y muerte cardiaca.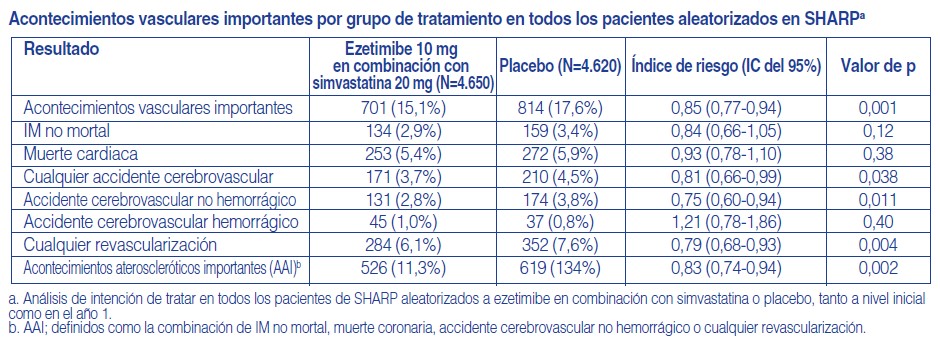
La reducción absoluta en el C-LDL alcanzada con ezetimibe en combinación con simvastatina fue menor entre los pacientes con un nivel inicial menor de C-LDL ( < 2,5 mmol/l) y en pacientes en diálisis al principio del estudio que en otros pacientes, y las reducciones del riesgo correspondientes en estos dos grupos se atenuaron.
Farmacocinética.
Después de la administración oral, ezetimibe es absorbido y conjugado extensamente a un glucurónido fenólico farmacológicamente activo (ezetimibe-glucurónido). Después de una sola dosis de 10 mg de ezetimibe, las concentraciones plasmáticas pico medias de ezetimibe (Cmax) de 3.4 a 5.5 ng/mL se alcanzaron en 4 a 12 horas (Tmax). Los valores medios de Cmax del ezetimibe-glucurónido de 45 a 71 ng/mL fueron alcanzados entre 1 y 2 horas (Tmax). No hubo desviación sustancial de la proporcionalidad de dosis entre 5 y 20 mg. La biodisponibilidad absoluta de ezetimibe no puede determinarse dado que el compuesto es virtualmente insoluble en el medio acuoso adecuado para la inyección. Ezetimibe tiene una biodisponibilidad variable; el coeficiente de variación, basado en la variabilidad inter-sujetos, fue de 35 a 60% para los valores de área bajo la curva (ABC). Efecto de los alimentos sobre la absorción: La administración concomitante de alimentos (grasos o no grasos) no tuvo efecto sobre la extensión de absorción de ezetimibe administrado como comprimidos de 10 mg. El valor de Cmax de ezetimibe aumentó un 38% con el consumo de alimentos muy grasos. Ezetimibe puede administrarse con o sin alimentos. Ezetimibe y el glucurónido de ezetimibe tienen una alta unión a las proteínas plasmáticas humanas ( > 90%). Ezetimibe es principalmente metabolizado en el intestino delgado y el hígado vía conjugación con glucurónido (reacción de fase II) con subsiguiente excreción biliar y renal. En todas las especies evaluadas se ha observado un metabolismo oxidativo mínimo (reacción de fase I). En humanos, ezetimibe es rápidamente metabolizada al glucurónido de ezetimibe. Ezetimibe y su glucurónido son los principales compuestos derivados de la droga detectados en plasma, constituyendo aproximadamente del 10 al 20% y del 80 al 90% de la droga total en plasma, respectivamente. Tanto ezetimibe como su glucurónido son lentamente eliminados del plasma con una vida media de aproximadamente 22 horas para ambos. Los perfiles concentración en plasma - tiempo muestran picos múltiples, lo que sugiere una importante circulación enterohepática. Con posterioridad a la administración oral de 14C-ezetimibe (20 mg) a seres humanos, el ezetimibe total (ezetimibe + ezetimibe-glucurónido) representó aproximadamente el 93% de la radioactividad total en plasma. Después de 48 horas, no hubo niveles detectables de radioactividad en el plasma. Entre 78% y 11% de la radioactividad administrada se recuperó en las heces y en la orina, respectivamente, a lo largo de un período de recolección de 10 días. Ezetimibe fue el principal componente en las heces y representó el 69% de la dosis administrada, mientras que el glucurónido de ezetimibe fue el principal componente en la orina y representó el 9% de la dosis administrada. Poblaciones Especiales: Pacientes Geriátricos: Posterior a la administración de dosis múltiples de 10 mg/día de ezetimibe durante 10 días, las concentraciones en plasma del ezetimibe total fueron casi dos veces más elevadas en sujetos sanos mayores (-65 años) en comparación con sujetos más jóvenes. Pacientes Pediátricos: Posterior a la administración de dosis múltiples de 10 mg/día de ezetimibe durante 7 días, la absorción y el metabolismo de ezetimibe fueron similares en adolescentes (10 a 18 años) y en adultos. No se dispone de datos farmacocinéticos en la población pediátrica en menores de 10 años de edad. Género: Posterior a la administración de dosis múltiples de 10 mg/día de ezetimibe durante 10 días, las concentraciones en plasma de ezetimibe total fueron levemente superiores ( < 20%) en las mujeres con respecto a los hombres. Raza: Basado en un meta-análisis de estudios farmacocinéticos de múltiples dosis, no se observaron diferencias farmacocinéticas entre la raza negra y la raza caucásica. Dado que muy pocos pacientes eran de otro grupo racial, no se han podido demostrar otras comparaciones farmacocinéticas. Insuficiencia Hepática: Después de una dosis única de 10 mg de ezetimibe, el área media bajo la curva (ABC) para ezetimibe total aumentó aproximadamente 1,7 veces en pacientes con insuficiencia hepática leve (Puntaje de Child-Pugh 5 a 6), en comparación con sujetos sanos. Los valores ABC medios para ezetimibe total y para ezetimibe aumentaron aproximadamente 3-4 veces y 5-6 veces, respectivamente, en pacientes con deterioro moderado (puntaje Child de Pugh 7 a 9) o severo (puntaje Child de Pugh 10 a 15). En un estudio de 14 días de duración, de múltiples dosis de ezetimibe 10 mg/día realizado en pacientes con insuficiencia hepática moderada los valores de ABC medios para ezetimibe total y para ezetimibe aumentaron aproximadamente 4 veces en el día 1 y en el día 14 en comparación con sujetos sanos. Debido a los efectos desconocidos de una mayor exposición a ezetimibe en pacientes con insuficiencia hepática moderada o severa, ezetimibe no es recomendado para estos pacientes (véanse Contraindicaciones y Precauciones, Insuficiencia Hepática). Insuficiencia Renal: Después de una dosis única de 10 mg de ezetimibe en pacientes con enfermedad renal severa (n=8; CrCl media 30 mL/min/1,73 m2), los valores ABC medios para ezetimibe total, el glucurónido de ezetimibe y ezetimibe se incrementaron aproximadamente 1,5 veces, en comparación con individuos sanos (n=9).
Indicaciones.
Hipercolesterolemia Primaria: CORACIL® está indicado, como monoterapia o asociado con una estatina, como tratamiento complementario de la dieta para la reducción del colesterol total (C-total), el colesterol-LDL (C-LDL) y las polipoproteínas B (Apo B) y triglicéridos y para el incremento del colesterol-HDL (C-HDL), en pacientes con hipercolesterolemia primaria (heterocigota familiar y no familiar). Hiperlipidemia mixta: CORACIL®, administrado en combinación con fenofibrato, está indicado como terapia adyuvante a la dieta para la reducción del C-total, C-LDL, Apo B, no C-HDL elevados, en pacientes con hiperlipidemia mixta. Hipercolesterolemia Familiar Homocigota (HFHo) CORACIL® está indicado asociado con una estatina para reducir el C-total y el C-LDL, como complemento de otros tratamientos hipolipemiantes (por ej.: Aféresis de LDL) o cuando estos tratamientos no se encuentren disponibles. Sitosterolemia homocigota: CORACIL® está indicado como tratamiento complementario de la dieta para reducir el sitosterol y el campesterol aumentado. El tratamiento con hipolipemiantes debe ser uno de los componentes de un tratamiento para reducir alguno de los múltiples factores de riesgo de enfermedad vascular ateroesclerótica por hipercolesterolemia. Los hipolipemiantes deben emplearse como complemento de una dieta apropiada (restricción de grasas saturadas y colesterol) y cuando la respuesta a la dieta y a otras medidas no farmacológicas ha sido inadecuada. (Guías ATP III). Antes de iniciar el tratamiento con CORACIL®, se deben descartar las causas secundarias de dislipidemia (diabetes, hipotiroidismo, enfermedad obstructiva hepática, insuficiencia renal crónica), y se deben considerar los fármacos que incrementen el C-LDL y/o disminuyan el C-HDL (progestágenos, esteroides anabólicos y corticosteroides), y tratar estas alteraciones si correspondiere o evaluar si corresponde suspender alguno de dichos fármacos. Es aconsejable realizar un perfil de lípidos para medir el C- total, el C-LDL, el C-HDL y TG. Para los niveles de TG > 400 mg/dL ( > 4,5 mmol/L), las concentraciones de C-LDL se deben determinar por ultracentrifugación. En el momento de la hospitalización por un evento coronario agudo, es aconsejable tomar las mediciones de lípidos en el momento de la admisión o dentro de las 24 horas. Estos valores pueden guiar al Médico para el inicio de un tratamiento hipolipemiante en el momento de la internación o de su alta.
Dosificación.
Los pacientes deben cumplir un régimen dietético para disminuir el colesterol antes de comenzar el tratamiento con CORACIL® y continuarlo mientras dure el tratamiento. La dosis recomendada es de 1 comprimido (10 mg) una vez por día. No se requiere ajuste de la dosis en pacientes con insuficiencia hepática leve, pacientes con insuficiencia renal o ancianos. CORACIL® puede administrarse con las comidas o lejos de ellas. Coadministración con estatinas o fenofibrato Se recomienda la toma de 1 comprimido de CORACIL® con estatinas o fenofibrato en el mismo momento del día y respetando las recomendaciones de dosificación de la medicación. Coadministración con secuestradores biliares Se recomienda administrar CORACIL® por lo menos 2 horas antes o 4 horas después de la administración del secuestrador biliar. Uso en pacientes pediátricos: El uso de ezetimibe en menores de 18 años no ha sido demostrado en pacientes con hipercolesterolemia familiar homocigota ni en pacientes con hiperlipidemia mixta. Niños y adolescentes -10 años: no se requieren ajustes de la dosis. Niños < 10 años: no se dispone de datos clínicos, por lo tanto, no se recomienda el tratamiento con CORACIL®.
Contraindicaciones.
Hipersensibilidad a alguno de los componentes de esta medicación. La combinación de CORACIL® con un inhibidor de HMG-CoA reductasa está contraindicada en pacientes con enfermedad hepática activa o elevaciones inexplicables y persistentes de las transaminasas séricas. Cuando CORACIL® es administrado junto con un inhibidor de HMG-CoA reductasa a una mujer con potencialidad de embarazarse, debe consultarse el prospecto del inhibidor de HMG-CoA reductasa específico. Todos los inhibidores de HMG-CoA reductasa están contraindicados en mujeres embarazadas o en período de lactancia. (Véase Precauciones, Embarazo).
Reacciones adversas.
Se definen las frecuencias como: muy frecuentes ≥1/10); frecuentes ≥1/100 a < 1/10); poco frecuentes ≥1/1.000 a < 1/100); raras ≥1/10.000 a < 1/1.000), muy raras ( < 1/10.000) y frecuencia no conocida (no puede estimarse a partir de los datos disponibles).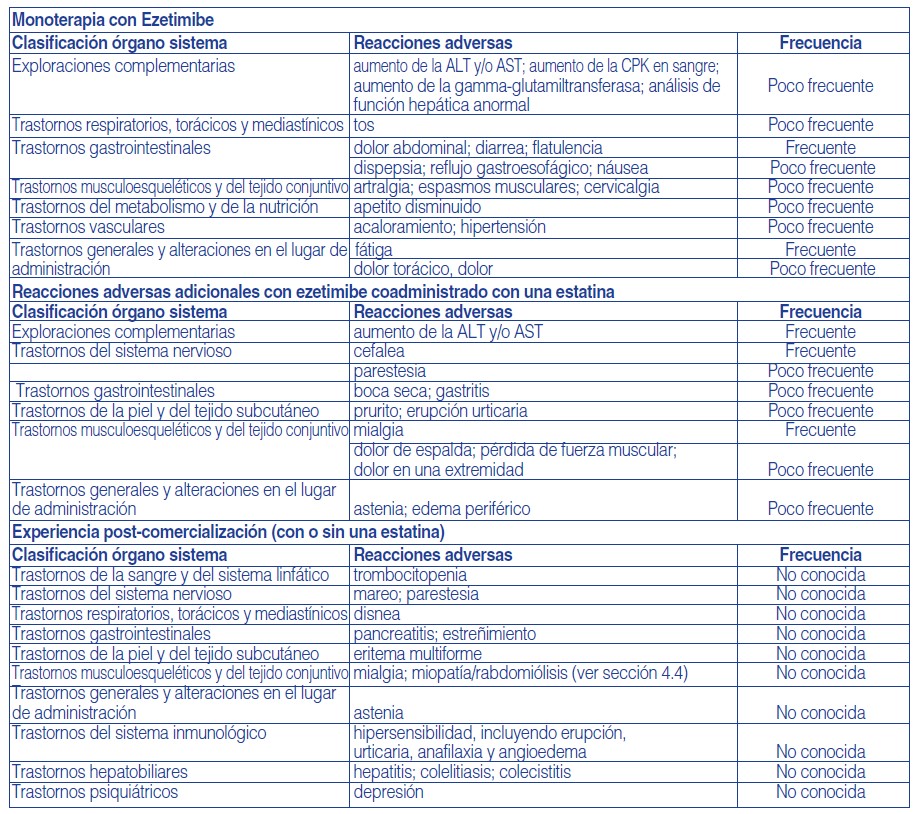
En el estudio IMPROVE-IT, realizado en 18.144 pacientes tratados con ezetimibe/simvastatina 10/40 mg (n=9.067; de los cuales al 6% se les incrementó la dosis a ezetimibe/simvastatina 10/80 mg) o con simvastatina 40 mg (n=9.077; de los cuales al 27% se les incrementó la dosis a simvastatina 80 mg), los perfiles de seguridad fueron similares durante una mediana de seguimiento de 6,0 años. La tasa de abandonos por acontecimientos adversos fue del 10,6% en los pacientes tratados con ezetimibe/simvastatina y del 10,1% en los pacientes tratados con simvastatina. La incidencia de miopatía fue del 0,2% con ezetimibe/simvastatina y del 0,1% con simvastatina. La miopatía se definió como debilidad o dolor muscular sin causa aparente con una concentración sérica de CPK - 10 x LSN o dos valores consecutivos de CPK - 5 y < 10 x LSN. La incidencia de rabdomiólisis fue del 0,1% con ezetimibe/simvastatina y del 0,2% con simvastatina, definiéndose rabdomiólisis como debilidad o dolor muscular sin causa aparente con una concentración sérica de CPK - 10 x LSN con lesión renal probada, dos valores consecutivos de CPK - 5 x LSN y < 10 x LSN con lesión renal probada o una CPK - 10.000 UI/l sin que se encuentre lesión renal. La incidencia de elevaciones consecutivas de las transaminasas (- 3 x LSN) fue del 2,5% con ezetimibe/simvastatina y del 2,3% con simvastatina. Se comunicaron efectos adversos relacionados con la vesícula biliar en el 3,1% de los pacientes tratados con ezetimibe/simvastatina y en el 3,5% de los que recibieron simvastatina. La incidencia de hospitalizaciones por colecistectomía fue del 1,5% en ambos grupos de tratamiento. Durante el estudio se diagnosticaron casos de cáncer (definidos como nuevos casos de cáncer) en el 9,4% frente al 9,5%, respectivamente. Pacientes con Enfermedad Renal Crónica: En el estudio "Study of Heart and Renal Protection" (SHARP), que incluyó a más de 9.000 pacientes tratados con una combinación a dosis fija de ezetimibe 10 mg con simvastatina 20 mg al día (n=4.650) o placebo (n=4.620), los perfiles de seguridad fueron comparables durante una mediana de seguimiento de 4,9 años. En este ensayo, sólo se registraron acontecimientos adversos graves e interrupciones del tratamiento debidos a cualquier acontecimiento adverso. Las tasas de interrupciones del tratamiento debidas a acontecimientos adversos fueron comparables (10,4% en pacientes tratados con ezetimibe en combinación con simvastatina, 9,8% en pacientes tratados con placebo). La incidencia de miopatía/rabdomiólisis fue del 0,2% en pacientes tratados con ezetimibe en combinación con simvastatina y del 0,1% en pacientes tratados con placebo. Las elevaciones consecutivas de las transaminasas ( > 3x LSN) se produjeron en el 0,7% de los pacientes tratados con ezetimibe en combinación con simvastatina, en comparación con el 0,6% de pacientes tratados con placebo. En este ensayo, no hubo aumentos estadísticamente significativos en la incidencia de acontecimientos adversos pre-especificados, incluyendo cáncer (9,4% con ezetimibe en combinación con simvastatina, 9,5% para placebo), hepatitis, colecistectomía o complicaciones de cálculos biliares o pancreatitis. Notificación de Sospecha de Reacciones Adversas: Es importante notificar la sospecha de reacciones adversas al medicamento tras su autorización. Ello permite una supervisión continuada de la relación beneficio/riesgo del medicamento. Se invita a los profesionales sanitarios a notificar las sospechas de reacciones adversas a través del Sistema Nacional de Farmacovigilancia al siguiente link: http://sistemas.anmat.gov.ar/aplicaciones_net/fvg_eventos_adversos_nuevo/index.htlm y/o al Departamento de Farmacovigilancia de GADOR S.A. vía email a farmacovigilancia@gador.com o telefónicamente al 0800-220-2273.
Precauciones.
La incidencia de aumento de las transaminasas ≥ 3 veces el límite superior normal) fue similar para ezetimibe (0,5%) que para placebo (0,3%). Se ha informado una mayor incidencia de aumento de las enzimas hepáticas con la asociación de Ezetimibe y estatinas (1,3%) que con la administración de la estatina sola (0,4%). Estos aumentos en general son asintomáticos, no se asocian con colestasis y retornan a los valores basales después de la interrupción del tratamiento o con la continuación del mismo. Se recomienda efectuar un control de la función hepática al iniciar el tratamiento y de acuerdo a lo recomendado para cada estatina específica. No se ha informado una mayor incidencia de miopatía o rabdomiólisis con Ezetimibe en comparación con el placebo o con las estatinas. Sin embargo, constituyen reacciones adversas conocidas de las estatinas y de otros hipolipemiantes por lo que deben ser tenidas en cuenta, especialmente cuando CORACIL® se administre asociado a otro hipolipemiante. La incidencia de las elevaciones de la CPK ( > 10 el límite superior normal) observada en estudios clínicos fue de 0,2% para ezetimibe y de 0,1% para placebo. Estas cifras fueron de 0,1% para ezetimibe más estatinas y de 0,4% para las estatinas como monoterapia. Se recomienda no administrar CORACIL® a pacientes con insuficiencia hepática moderada a severa. (Véase Poblaciones especiales). Interacciones con otras drogas: Colestiramina: la administración concomitante de colestiramina (4 g dos veces al día) disminuyó los valores medios de ABC del ezetimibe total y del ezetimibe aproximadamente un 55% y 80% respectivamente. La reducción incremental de C-LDL que debería observarse por el agregado de ezetimibe a la colestiramina puede disminuir por esta interacción. Fibratos: La co-administración de ezetimibe con fibratos (distintos al fenofibrato) no ha sido estudiada. Los fibratos pueden incrementar la excreción de colesterol en la bilis y producir una colelitiasis. No se recomienda la coadministración de ezetimibe con fibratos (con la excepción del fenofibrato) hasta que se estudie su uso en pacientes. Fenofibrato: En un estudio farmacocinético la administración concomitante de fenofibrato incrementó las concentraciones de ezetimibe total aproximadamente 1,5 veces. La farmacocinética del fenofibrato no fue afectada significativamente por ezetimibe 10 mg/día. Ante la sospecha de colelitiasis en un paciente que está recibiendo ezetimibe y fenofibrato, debe indicarse un estudio de vesícula biliar y un tratamiento alternativo para disminuir los niveles lipídicos debe ser considerado. Gemfibrozil: En un estudio farmacocinético la administración concomitante de gemfibrozil incrementó las concentraciones de ezetimibe total aproximadamente 1,7 veces. Inhibidores de HMG-CoA reductasa: No se observaron interacciones farmacocinéticas clínicamente significativas cuando ezetimibe se coadministró con atorvastatina, simvastatina, pravastatina, lovastatina, fluvastatina o rosuvastatina. Ciclosporina: Se debe tener precaución al iniciar un tratamiento con ezetimibe en pacientes tratados con ciclosporina debido al aumento en las concentraciones de ezetimibe. Este aumento puede ser mayor en pacientes con insuficiencia renal severa. En pacientes que reciban en forma concomitante ambas medicaciones las consecuencias de la reducción de los niveles lipídicos como resultado de este aumento de las concentraciones de ezetimibe debe ser especialmente tenido en cuenta. Los pacientes que reciben ezetimibe junto con ciclosporina deben ser cuidadosamente monitoreados. Warfarina: La administración concomitante de ezetimibe (10 mg una vez al día) no tuvo efecto significativo sobre la biodisponibilidad de arfarina y el tiempo de protrombina en un estudio realizado en doce hombres adultos sanos. Digoxina: La administración concomitante de ezetimibe (10 mg una vez al día) no tuvo efecto significativo sobre la biodisponibilidad de digoxina ni sobre los parámetros del ECG (FC, intervalos PR, QT y QTc) en un estudio realizado en doce hombres adultos sanos. Anticonceptivos Orales: La coadministración de ezetimibe (10 mg una vez al día) con anticonceptivos orales no tuvo efecto significativo sobre la biodisponibilidad de etinilestradiol o levonorgestrel en un estudio en 18 mujeres adultas sanas. Cimetidina: Dosis múltiples de cimetidina (400 mg dos veces al día) no tuvieron efecto significativo sobre la biodisponibilidad oral de ezetimibe y ezetimibe total en un estudio realizado en doce adultos sanos Antiácidos: En un estudio en 12 adultos sanos, la administración de una dosis única de antiácido (hidróxido de aluminio + hidróxido de magnesio) no tuvo efecto significativo sobre la biodisponibilidad oral de ezetimibe total, del glucurónido de ezetimibe, ni de ezetimibe en los valores de ABC. El valor de Cmáx de ezetimibe total disminuyó en un 30%. Glipizida: En un estudio en 12 hombres adultos sanos, los niveles de ezetimibe en condiciones estables (10 mg una vez al día) no tuvieron efecto significativo sobre la farmacocinética y la farmacodinamia de glipizida. Una sola dosis de glipizida (10 mg) no tuvo efecto significativo sobre la exposición a ezetimibe total o a ezetimibe. En un estudio realizado en doce hombres adultos sanos, ezetimibe no tuvo efecto significativo sobre una serie de drogas que son metabolizadas por el citocromo P450 (1ª2, 2D6, 2C8/9 y 3ª4) (cafeína, dextrometorfan, tolbutamida y midazolam IV). Esto indica que ezetimibe no es ni un inhibidor ni un inductor de estas isoenzimas del citocromo P450 y es improbable que ezetimibe afecte el metabolismo de drogas que son metabolizadas por estas enzimas. Carcinogénesis: Se llevó a cabo un estudio dietario sobre carcinogenicidad de 104 semanas con ezetimibe en ratas, en dosis de hasta 1500 mg/kg/día (machos) y 500 mg/kg/día (hembras) (aproximadamente 20 veces la exposición humana de 10 mg diarios, basados en ABC 0-24hs para el ezetimibe total). Un estudio dietario sobre carcinogenicidad de 104 semanas con ezetimibe también se llevó a cabo en ratones en dosis de hasta 500 mg/kg/día (más de 150 veces la exposición humana a los 10 mg diarios basados en ABC 0-24hs para el ezetimibe total). No hubo incrementos estadísticamente significativos en las incidencias de tumores en ratas o ratones tratados con el fármaco. Mutagénesis: No se observaron evidencias de mutagenicidad en un test de mutagenicidad microbiana in vitro (Ames) con Salmonella typhimurium y Escherichia coli con o sin activación metabólica. No se observaron evidencias de clastogenicidad en un ensayo de aberración cromosómica in vitro en linfocitos humanos de sangre periférica con activación metabólica o sin ella. Sumado a ello, no hubo evidencias de genotoxicidad en el test del micronúcleo en ratones in vivo. Embarazo y Reproducción: No existen estudios adecuados y bien controlados de ezetimibe en mujeres embarazadas. Ezetimibe solo se debe usar durante el embarazo en el caso que el beneficio potencial justifique el riesgo para el feto. En estudios por vía oral de ezetimibe sobre el desarrollo embrio-fetal llevados a cabo en ratas y conejos durante la organogénesis, no hubo evidencias de efectos embrioletales a las dosis estudiadas (250, 500, 1000 mg/kg/día). En ratas, se observó una mayor incidencia de hallazgos esqueléticos fetales comunes (par extra de costillas torácicas, centros espinales de las vértebras cervicales sin osificar, costillas más cortas) con 1000 mg/kg/día (aproximadamente 10 veces la exposición humana a los 10 mg diarios basados en ABC 0-24 hs para ezetimibe total). En los conejos tratados con ezetimibe se observó una mayor incidencia de costillas torácicas extra con 1000 mg/kg/día (150 veces la exposición humana a los 10 mg diarios basados en ABC0-24 hs para ezetimibe total). Ezetimibe atravesó la placenta cuando las ratas y conejas preñadas recibieron dosis orales múltiples. Estudios con dosis múltiples de ezetimibe administrado en combinación con inhibidores de la HMG-CoA reductasa en ratas y conejos durante la organogénesis dieron por resultado mayores exposiciones a ezetimibe y a las estatinas. Los hallazgos a nivel de reproducción se presentaron con dosis más bajas en el tratamiento combinado comparado con la monoterapia. Todos los inhibidores de la HMG-CoA reductasa están contraindicados en mujeres embarazadas ó en período de lactancia. Cuando CORACIL® es administrado junto con un inhibidor de HMG-CoA reductasa a una mujer que puede quedar embarazada, se debe consultar el prospecto de ese inhibidor de la HMG-CoA reductasa. (Véase Contraindicaciones). En los estudios de fertilidad de ezetimibe por vía oral llevados a cabo en ratas, no hubo evidencias de toxicidad reproductiva en dosis de hasta 1000 mg/kg/día en ratas macho o hembra (aproximadamente 7 veces la exposición humana a los 10 mg diarios basados en ABC0-24hs para ezetimibe total). Lactancia: No se sabe si ezetimibe se excreta en la leche humana; por lo tanto CORACIL® no se debe usar en madres en período de lactancia a menos que el beneficio potencial justifique el riesgo para el lactante. Empleo en pediatría: La farmacocinética de ezetimibe en adolescentes (10 a 18 años) ha demostrado ser similar a la de los adultos. La experiencia en el tratamiento con ezetimibe en la población pediátrica se limita a 4 pacientes (9 a 17 años) en el estudio de sitosterolemia y a 5 pacientes (11 a 17 años) en el estudio de pacientes con hipercolesterolemia familiar homocigota. No se recomienda el tratamiento con CORACIL® en niños < 10 años. Empleo en geriatría: De los pacientes que recibieron ezetimibe en los estudios clínicos, 948 tenían 65 años o más (esto incluyó 206 con 75 años y mayores). La efectividad e inocuidad de ezetimibe fue similar entre estos pacientes e individuos más jóvenes. No se puede descartar la mayor sensibilidad de algunas personas mayores.
Advertencias.
La administración conjunta de ezetimibe con una estatina o con fenofibrato debe estar de acuerdo a lo indicado en el prospecto de estos últimos. Enzimas hepáticas En ensayos clínicos controlados en los que se administró ezetimibe junto a una estatina, se observaron elevaciones consecutivas de las transaminasas (3 x límite superior normal [LSN]). Cuando se administre CORACIL® junto a una estatina, deben realizarse pruebas de función hepática al inicio del tratamiento y seguir las recomendaciones de la estatina a este respecto En el tudio IMPROVE-IT (IMProved Reduction of Outcomes: Vytorin Efficacy International Trial), 18.144 pacientes con cardiopatía coronaria y antecedentes de un acontecimiento de SCA fueron aleatorizados para recibir ezetimibe/simvastatina 10/40 mg al día (n=9.067) o simvastatina 40 mg al día (n=9.077). Con una mediana de seguimiento de 6,0 años, la incidencia de elevaciones consecutivas de las transaminasas (3 x LSN) fue del 2,5% para ezetimibe/simvastatina y del 2,3% para simvastatina. En un ensayo clínico controlado en el que más de 9.000 pacientes con enfermedad renal crónica fueron aleatorizados para recibir ezetimibe 10 mg en combinación con simvastatina 20 mg al día (n=4.650) o placebo (n=4.620) (mediana de seguimiento de 4,9 años), la incidencia de elevaciones consecutivas de las transaminasas ( > 3 x LSN) fue del 0,7% para ezetimibe en combinación con simvastatina y del 0,6 % para placebo. Sistema musculoesquelético: En la experiencia post-comercialización con ezetimibe, se han comunicado casos de miopatía y rabdomiólisis. La mayoría de los pacientes que desarrollaron rabdomiólisis tomaban una estatina concomitantemente con ezetimibe. Sin embargo, se han comunicado muy raramente casos de rabdomiólisis con ezetimibe en monoterapia y muy raramente con la adición de ezetimibe a otros fármacos que aumentan el riesgo de rabdomiólisis. Si se sospecha miopatía en base a los síntomas musculares o si se confirma por un nivel de la creatinina fosfoquinasa (CPK) > 10 veces el límite superior normal, CORACIL®, cualquier estatina y cualquiera de estos otros fármacos que el paciente esté tomando concomitantemente deben interrumpirse inmediatamente. Todos los pacientes que empiecen el tratamiento con CORACIL® deben ser advertidos del riesgo de miopatía y que deben informar rápidamente si aparece dolor, sensibilidad a la presión o debilidad muscular sin causa aparente. En el estudio IMPROVE-IT, 18.144 pacientes con cardiopatía coronaria y antecedentes de un acontecimiento de SCA fueron aleatorizados para recibir ezetimibe/simvastatina 10/40 mg al día (n=9.067) o simvastatina 40 mg al día (n=9.077). Con una mediana de seguimiento de 6,0 años, la incidencia de miopatía fue del 0,2% para ezetimibe/simvastatina y del 0,1% para simvastatina. La miopatía se definió como debilidad o dolor muscular sin causa aparente con una concentración sérica de CPK > 10 x LSN o dos valores consecutivos de CPK - 5 y < 10 x LSN. La incidencia de rabdomiólisis fue del 0,1% para ezetimibe/simvastatina y del 0,2% para simvastatina, definiéndose rabdomiólisis como debilidad o dolor muscular sin causa aparente con una concentración sérica de CPK > 10 x LSN con lesión renal probada, dos valores consecutivos de CPK - 5 x LSN y < 10 x LSN con lesión renal probada o CPK -10.000 UI/l sin que se encuentre lesión renal. En un ensayo clínico controlado en el que más de 9.000 pacientes con enfermedad renal crónica fueron aleatorizados para recibir ezetimibe 10 mg en combinación con simvastatina 20 mg al día (n=4.650) o placebo (n=4.620) (mediana de seguimiento de 4,9 años), la incidencia de miopatía/rabdomiólisis fue del 0,2% para ezetimibe en combinación con simvastatina y del 0,1% para placebo. Pacientes con deterioro hepático: Dado que no se conocen los efectos del aumento de la exposición a ezetimibe en los pacientes con deterioro hepático moderado o grave, no se recomienda la administración de CORACIL®. Este medicamento contiene lactosa Los pacientes con intolerancia hereditaria a galactosa, insuficiencia de lactasa de Lapp (insuficiencia observada en ciertas poblaciones de Laponia) o malabsorción de glucosa o galactosa no deben tomar este medicamento.
Conservación.
Conservar a temperatura ambiente. Proteger de la humedad.
Sobredosificación.
En el caso de una sobredosis se deberán emplear medidas sintomáticas y de apoyo. Muy pocos casos de sobredosificación con ezetimibe han sido informados. Estos no han sido asociados a experiencias adversas serias. Ante la eventualidad de una sobredosificación, concurrir al Hospital más cercano o comunicarse con los Centros de Toxicología: Hospital de Pediatría Ricardo Gutiérrez - Buenos Aires: (011) 4962-6666/2247 Hospital Nacional Alejandro Posadas - Buenos Aires: (011) 4654-6648/4658-7777 Optativamente otros Centros de Intoxicaciones.
Presentación.
Envases conteniendo 10 y 30 comprimidos.
Revisión.
10/2016. G00073700-08.