VIREAD®
GADOR
Antirretroviral contra el HIV.
Composición.
Cada comprimido recubierto de VIREAD® contiene: Tenofovir disoproxil fumarato (equivalente a 245 mg de tenofovir disoproxil 300 mg. Excipientes: Almidón pregelatinizado, Croscarmelosa sódica, Lactosa monohidrato, Celulosa microcristalina, Estearato de magnesio, Opadry ll Y-3010671 A* c.s. * laca alumínica FD&C Azul No 2, hipromelosa 2910, monohidrato de lactosa, dióxido de titanio y triacetina.
Farmacología.
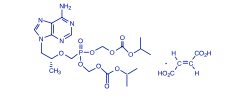
Descripción: VIREAD® es el nombre comercial del tenofovir DF (un profármaco de tenofovir), que es una sal del ácido fumárico del éster bis-isopropoxicarboniloximetilo derivado de tenofovir. In vivo, el tenofovir DF se convierte en tenofovir, análogo fosfonato nucleósido acíclico (nucleótido) de 5'-monofosfato de adenosina. El tenofovir muestra actividad contra la retrotranscriptasa del VIH-1. El nombre químico del tenofovir disoproxil fumarato es fumarato de 9-[(R)-2-[[bis[[(isopropoxicarbonil)oxi]metoxi]fosfinil]metoxi]propil]adenina (1:1). Su fórmula molecular es C19H30N5O10P•C C4H4O4, y su peso molecular, 635,52. Tiene la siguiente fórmula estructural:
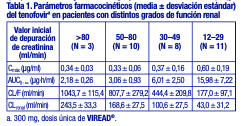
El tenofovir DF es un polvo cristalino, de color blanco a blanquecino, con una solubilidad de aproximadamente 13,4 mg/ml en agua destilada a 25 °C. Tiene un coeficiente de partición (log p) del amortiguador de octanol/ fosfato (pH 6,5) de 1,25 a 25 °C. Los comprimidos VIREAD® son para administración oral. Cada comprimido contiene 300 mg de tenofovir DF, lo que es equivalente a 245 mg de disoproxilo de tenofovir, y además contiene los siguientes excipientes: croscaramelosa sódica, lactosa monohidrato, estearato de magnesio, celulosa microcristalina y almidón pregelatinizado. Los comprimidos están recubiertos con Opadry II Y-30-10671-A, que contiene laca Alumínica FD&C Azul No. 2, hipromelosa 2910, monohidrato de lactosa, dióxido de titanio y triacetina. En este prospecto, todas las dosis se expresan en términos de tenofovir DF, excepto cuando se indique lo contrario. Farmacología clínica: Mecanismo de acción: El tenofovir DF es un antiviral [véase Microbiología]. Farmacocinética: Se han evaluado las propiedades farmacocinéticas del tenofovir DF en voluntarios sanos y en personas infectadas con el VIH-1. Las propiedades farmacocinéticas del tenofovir son similares en estas poblaciones. Absorción: VIREAD® es un profármaco diéster hidrosoluble del principio activo tenofovir. La biodisponibilidad oral del tenofovir en VIREAD® en los pacientes en ayunas es de aproximadamente el 25 %. Después de la administración en ayunas por vía oral de una dosis única de VIREAD® de 300 mg a pacientes infectados con el VIH-1, las concentraciones máximas (Cmáx) en el suero se alcanzan en 1,0 ± 0,4 h. El valor de la Cmáx es de 0,30 ± 0,09mg/ml, y el del AUC de 2,29 ± 0,69mg•h/ml. Las propiedades farmacocinéticas del tenofovir son proporcionales a la dosis en un intervalo de dosis de VIREAD® de 75 a 600 mg, y no se ven afectadas por las dosis repetidas. Distribución: La unión in vitro del tenofovir a las proteínas en el plasma humano es inferior al 0,7 %, y en el suero humano, al 7,2 %, en el intervalo de concentración de tenofovir de 0,01 a 25mg/ml. El volumen de distribución en estado de equilibrio es de 1,3 ± 0,6 l/kg después de una administración intravenosa de tenofovir de 1,0 mg/kg, y de 1,2 ± 0,4 l/kg después de una administración de 3,0 mg/kg. Metabolismo y eliminación: Los estudios in vitro indican que ni el disoproxilo de tenofovir ni el tenofovir son sustratos de las enzimas CYP. Después de la administración intravenosa de tenofovir, aproximadamente entre el 70 y el 80 % de la dosis se recupera en la orina como tenofovir inalterado en las 72 horas siguientes. Después de la administración oral de una dosis única de VIREAD®, la semivida de eliminación terminal del tenofovir es de aproximadamente 17 horas. Después de la administración de múltiples dosis orales de VIREAD® de 300 mg una vez por día (con alimentos), el 32 ± 10 % de la dosis administrada se recupera en la orina en el lapso de 24 horas. El tenofovir se elimina mediante una combinación de filtración glomerular y secreción tubular activa. Puede haber competencia por la eliminación con otros compuestos que también se eliminan por vía renal. Efectos de los alimentos en la absorción oral: La administración de los comprimidos de 300 mg de VIREAD® después de una comida con un alto contenido graso (semejante a 700 a 1000 kcal con un contenido graso del 40 al 50 %) aumenta la biodisponibilidad oral, con un aumento del AUC0-inf del tenofovir de aproximadamente el 40 %, y un aumento en la Cmáx de aproximadamente el 14 %. Sin embargo, la administración de VIREAD® con una comida ligera no tiene efectos significativos en las propiedades farmacocinética del tenofovir si se compara con la administración del fármaco en ayunas. Los alimentos retrasan el tiempo para alcanzar la Cmáx de tenofovir en aproximadamente una hora. La Cmáx del tenofovir es de 0,33 ± 0,12mg/ml, y el AUC, 3,32 ± 1,37mg•h/ ml, después de múltiples dosis de VIREAD® de 300 mg, una vez al día, con alimentos, sin controlar el contenido de estos últimos. Poblaciones especiales: Raza: No hubo un número suficiente de grupos raciales y étnicos diferentes a la raza blanca para poder determinar adecuadamente las diferencias farmacocinéticas entre estas poblaciones. Sexo: Las propiedades farmacocinéticas del tenofovir son similares en los pacientes varones y mujeres. Pacientes pediátricos de 12 años o más: Las propiedades farmacocinéticas del tenofovir en estado de equilibrio se evaluaron en 8 pacientes pediátricos infectados por el VIH 1 (de 12 a menos de 18 años de edad). Los valores medios (±DE) de Cmáx y AUCtau son 0,38 ± 0,13mg/ml y 3,39 ± 1,22mg•h/ml, respectivamente. La exposición al tenofovir alcanzada en estos pacientes pediátricos que recibieron dosis diarias orales de 300 mg de VIREAD® fue similar a las exposiciones alcanzadas en adultos que recibieron dosis únicas diarias de 300 mg de VIREAD®. Las exposiciones al tenofovir observadas en 52 pacientes pediátricos infectados por el VHB (de 12 a menos de 18 años de edad) que recibieron dosis únicas diarias de 300 mg de VIREAD® por vía oral fueron comparables a las exposiciones alcanzadas en adultos y adolescentes infectados por el VIH-1 que recibieron dosis diarias únicas de 300 mg. No se han realizado estudios farmacocinéticos en pacientes pediátricos menores de 12 años. Pacientes geriátricos: No se han realizado estudios farmacocinéticos en ancianos (65 años o más). Pacientes con disfunción renal: Las propiedades farmacocinéticas del tenofovir están alteradas en los pacientes con disfunción renal [véase Advertencias]. En los pacientes con una depuración de creatinina inferior a 50 ml/min o con enfermedad renal terminal (ERT) que requieren diálisis, la Cmáx y el AUC0-inf del tenofovir aumentaron (tabla 1). Se recomienda modificar el intervalo de dosificación de VIREAD® en los pacientes con una depuración de creatinina estimada inferior a 50 ml/min o en los pacientes con enfermedad renal terminal que precisan diálisis [véase Dosificación].
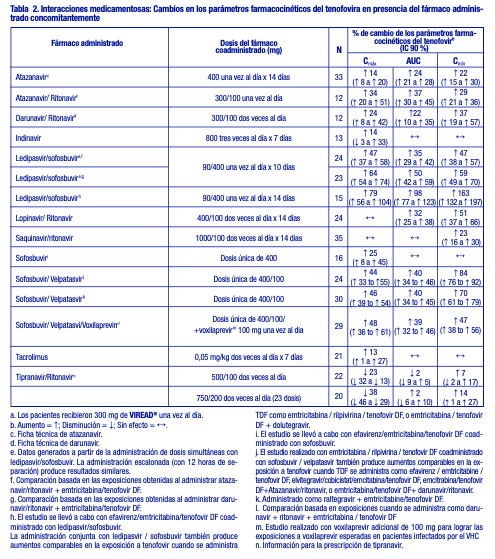
El tenofovir se elimina de forma eficaz mediante hemodiálisis, con un coeficiente de extracción de aproximadamente el 54 %. Después de una dosis única de 300 mg de VIREAD®, una sesión de hemodiálisis de cuatro horas eliminó aproximadamente el 10 % de la dosis de tenofovir administrada. Pacientes con disfunción hepática: Se han estudiado las propiedades farmacocinéticas del tenofovir después de una dosis única de 300 mg de VIREAD® en pacientes no infectados por el VIH y con disfunción hepática de moderada a grave. No hubo alteraciones importantes en las propiedades farmacocinéticas del tenofovir en pacientes con disfunción hepática, en comparación con los pacientes con una función hepática normal. No se requiere ningún cambio de la dosificación de VIREAD® en los pacientes con disfunción hepática. Evaluación de las interacciones medicamentosas: En concentraciones considerablemente mayores (~300 veces) que las observadas in vivo, el tenofovir no inhibió el metabolismo del fármaco in vitro mediado por ninguna de las siguientes isoformas CYP en los seres humanos: CYP3A4, CYP2D6, CYP2C9 o CYP2E1. Sin embargo, se observó una disminución pequeña (6 %), pero estadísticamente significativa, en el metabolismo del sustrato de CYP1A. Sobre la base de los resultados de los experimentos in vitro y la vía de eliminación conocida del tenofovir, la posibilidad de interacciones mediadas por el CYP que afectan al tenofovir con otros medicamentos es baja. Se ha evaluado VIREAD® en voluntarios sanos en asociación con otros antirretrovirales y posibles fármacos concomitantes. Las tablas 2 y 3 resumen los efectos farmacocinéticos del fármaco administrado concomitantemente sobre las propiedades farmacocinéticas del tenofovir, y los efectos de VIREAD® sobre las propiedades farmacocinéticas del fármaco coadministrado. El TDF es un sustrato de los transportadores de la P-glicoproteína (P-gp) y de la proteína de resistencia al cáncer de mama (BCRP). Cuando el TDF se administra conjuntamente con un inhibidor de estos transportadores, se puede observar un aumento en la absorción. No se han observado interacciones medicamentosas clínicamente significativas entre VIREAD® y el efavirenz, la metadona, el nelfinavir, los anticonceptivos orales, la ribavirina o el sofosbuvir. Ver tabla 2.
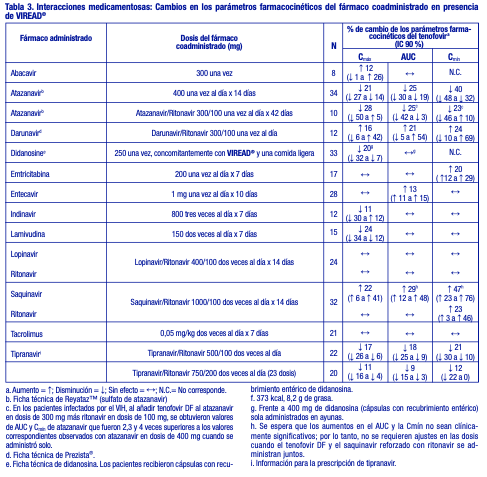
No se observó ningún efecto en los parámetros farmacocinéticos de los siguientes fármacos coadministrados con VIREAD®: abacavir, didanosina (comprimidos amortiguados), emtricitabina, entecavir y lamivudina. Ver tabla 3.
Microbiología: Mecanismo de acción: El tenofovir DF es un diéster de fosfonato nucleosídico acíclico análogo del monofosfato de adenosina. El tenofovir DF requiere la hidrólisis inicial del diéster para su conversión a tenofovir y fosforilaciones subsiguientes por medio de enzimas celulares para formar el difosfato de tenofovir, un terminador de cadenas obligado. El difosfato de tenofovir inhibe la actividad de la retrotranscriptasa del VIH-1 y de la retrotranscriptasa del VHB al competir con el sustrato natural de 5'-trifosfato de desoxiadenosina y, después de la incorporación al ADN, por la terminación de la cadena de ADN. El difosfato de tenofovir es un inhibidor débil de las polimerasas del ADN de mamíferos a, b, de la polimerasa del ADN mitocondrial c. Actividad contra el VIH Actividad antiviral: Se evaluó la actividad antiviral del tenofovir contra las cepas aisladas clínicas y de laboratorio de VIH-1 en las líneas celulares linfoblastoides, células monocito-macrofágicas primarias y linfocitos sanguíneos periféricos. Los valores de concentración eficaz al 50 % (CE50) del tenofovir estuvieron en el intervalo entre 0,04 mM y 8,5 mM. En los estudios de asociación de fármacos, tenofovir no fue antagonista de los inhibidores nucleosídicos de la retrotranscriptasa (abacavir, didanosina, lamivudina, estavudina, zalcitabina, zidovudina), los inhibidores no nucleosídicos de la retrotranscriptasa (delavirdina, efavirenz, nevirapina) ni los inhibidores de la proteasa (amprenavir, indinavir, nelfinavir, ritonavir, saquinavir). El tenofovir mostró actividad antiviral en cultivo celular contra los subtipos A, B, C, D, E, F, G y O del VIH-1 (los valores de CE50 variaron entre 0,5 mM y 2,2 mM) y actividad específica de la cepa contra el VIH-2 (los valores de CE50 variaron entre 1,6 mM y 5,5 mM). Resistencia: Se han seleccionado en un cultivo celular cepas aisladas del VIH-1 con una disminución de la sensibilidad al tenofovir. Estos virus expresaron una sustitución K65R en la retrotranscriptasa y mostraron una reducción de 2 a 4 veces en la sensibilidad al tenofovir. Además, una sustitución K70E fue seleccionada por el tenofovir en la transcriptasa inversa del VIH 1, y genera una sensibilidad reducida de bajo nivel al tenofovir. En el estudio 903 de pacientes sin tratamiento previo con antirretrovirales (VIREAD® + lamivudina + efavirenz, en comparación con estavudina + lamivudina + efavirenz) [véase Estudios clínicos (8.1)], los análisis genotípicos de cepas aisladas de pacientes con fracaso virológico después de 144 semanas demostraron que el desarrollo de sustituciones asociadas con la resistencia al efavirenz y a la lamivudina era muy frecuente y sin diferencias entre los grupos de tratamiento. La sustitución K65R se presentó en 8/47 (17 %) de las cepas aisladas de los pacientes analizados en el grupo tratado con VIREAD®, y en 2/49 (4 %) de las cepas aisladas de los pacientes analizados en el grupo tratado con estavudina. De los 8 pacientes cuyo virus desarrolló K65R en el grupo tratado con VIREAD® después de 144 semanas de tratamiento, 7 de los casos ocurrieron en las primeras 48 semanas de tratamiento, y uno a las 96 semanas. Un paciente en el grupo de VIREAD® desarrolló la sustitución K70E en el virus. En este estudio no se identificaron otras sustituciones que dieran como resultado resistencia a VIREAD®. En el estudio 934 de pacientes sin tratamiento previo con antirretrovirales (VIREAD® + EMTRIVA® + efavirenz, en comparación con zidovudina (AZT)/lamivudina (3TC) + efavirenz) [véase Estudios clínicos], los análisis genotípicos realizados en cepas aisladas del VIH-1 de todos los pacientes con fracaso virológico confirmado con más de 400 copias/ml de ARN del VIH-1 en la semana 144 o que suspendieron el tratamiento de forma prematura demostraron que el desarrollo de sustituciones asociadas con la resistencia al efavirenz era muy frecuente y similar entre los dos grupos de tratamiento. Se observó la sustitución de aminoácidos M184V, asociada con resistencia a EMTRIVA® y lamivudina, en 2/19 de las cepas aisladas de los pacientes analizados en el grupo tratado con VIREAD® + EMTRIVA®, y en 10/29 de las cepas aisladas de los pacientes analizados en el grupo tratado con zidovudina/lamivudina. En las 144 semanas del estudio 934, ningún paciente había presentado una sustitución K65R detectable en su VIH-1, según los análisis genotípicos habituales. Resistencia cruzada: Se ha identificado resistencia cruzada entre ciertos inhibidores de la retrotranscriptasa. Las sustituciones de K65R y K70E seleccionadas por el tenofovir también están seleccionadas en algunos pacientes infectados por el VIH-1 tratados con abacavir o didanosina. Las cepas aisladas del VIH-1 con esta sustitución también muestran una disminución de la sensibilidad a la emtricitabina y a la lamivudina. Por lo tanto, puede presentarse resistencia cruzada entre estos fármacos en pacientes cuyos virus hospedan la sustitución K65R o K70E. Las cepas aisladas del VIH-1 de los pacientes (N = 20) cuyo VIH-1 expresó un promedio de tres sustituciones de la retrotranscriptasa asociadas a la zidovudina (M41L, D67N, K70R, L210W, T215Y/F o K219Q/E/N) mostraron una disminución de 3,1 veces de la sensibilidad al tenofovir. En los estudios 902 y 907 realizados en pacientes con tratamiento previo con antirretrovirales (VIREAD® + tratamiento de fondo estándar (TFE), en comparación con placebo + TFE) [véase Estudios clínicos (8.1)], 14/304 (5 %) de los pacientes tratados con VIREAD® con fracaso virológico hasta la semana 96 tuvieron una disminución superior a 1,4 veces (mediana, 2,7 veces) de la sensibilidad al tenofovir. El análisis genotípico de las cepas aisladas iniciales y de fracaso mostró presentación de la sustitución K65R en el gen de la retrotranscriptasa del VIH-1. Se ha evaluado la respuesta virológica al tratamiento con VIREAD® con respecto al genotipo viral inicial (N = 222) en pacientes con tratamiento previo con antirretrovirales que participaron en los estudios 902 y 907. En estos estudios clínicos, el 94 % de los participantes evaluados tenían cepas aisladas del VIH-1 iniciales que expresaban al menos una sustitución para el inhibidor nucleosídico de la retrotranscriptasa (INRT). Las respuestas virológicas de los pacientes del subestudio de genotipos fueron similares a los resultados generales del estudio. Se realizaron varios análisis exploratorios para evaluar el efecto de sustituciones específicas y pautas mutacionales en los resultados virológicos. Debido al gran número de comparaciones potenciales, no se realizaron pruebas estadísticas. Se observaron varios grados de resistencia cruzada de VIREAD® a sustituciones asociadas con la resistencia a la zidovudina preexistentes (M41L, D67N, K70R, L210W, T215Y/F o K219Q/E/N) y parecían depender del tipo y el transcriptasa inversa del VIH 1, y genera una sensibilidad reducida de bajo nivel al tenofovir. En el estudio 903 de pacientes sin tratamiento previo con antirretrovirales (VIREAD® + lamivudina + efavirenz, en comparación con estavudina + lamivudina + efavirenz) [véase Estudios clínicos], los análisis genotípicos de cepas aisladas de pacientes con fracaso virológico después de 144 semanas demostraron que el desarrollo de sustituciones asociadas con la resistencia al efavirenz y a la lamivudina era muy frecuente y sin diferencias entre los grupos de tratamiento. La sustitución K65R se presentó en 8/47 (17 %) de las cepas aisladas de los pacientes analizados en el grupo tratado con VIREAD®, y en 2/49 (4 %) de las cepas aisladas de los pacientes analizados en el grupo tratado con estavudina. De los 8 pacientes cuyo virus desarrolló K65R en el grupo tratado con VIREAD® después de 144 semanas de tratamiento, 7 de los casos ocurrieron en las primeras 48 semanas de tratamiento, y uno a las 96 semanas. Un paciente en el grupo de VIREAD® desarrolló la sustitución K70E en el virus. En este estudio no se identificaron otras sustituciones que dieran como resultado resistencia a VIREAD®. En el estudio 934 de pacientes sin tratamiento previo con antirretrovirales (VIREAD® + EMTRIVA® + efavirenz, en comparación con zidovudina (AZT)/lamivudina (3TC) + efavirenz) [véase Estudios clínicos], los análisis genotípicos realizados en cepas aisladas del VIH-1 de todos los pacientes con fracaso virológico confirmado con más de 400 copias/ml de ARN del VIH-1 en la semana 144 o que suspendieron el tratamiento de forma prematura demostraron que el desarrollo de sustituciones asociadas con la resistencia al efavirenz era muy frecuente y similar entre los dos grupos de tratamiento. Se observó la sustitución de aminoácidos M184V, asociada con resistencia a EMTRIVA® y lamivudina, en 2/19 de las cepas aisladas de los pacientes analizados en el grupo tratado con VIREAD® + EMTRIVA®, y en 10/29 de las cepas aisladas de los pacientes analizados en el grupo tratado con zidovudina/lamivudina. En las 144 semanas del estudio 934, ningún paciente había presentado una sustitución K65R detectable en su VIH-1, según los análisis genotípicos habituales. Resistencia cruzada: Se ha identificado resistencia cruzada entre ciertos inhibidores de la retrotranscriptasa. Las sustituciones de K65R y K70E seleccionadas por el tenofovir también están seleccionadas en algunos pacientes infectados por el VIH-1 tratados con abacavir o didanosina. Las cepas aisladas del VIH-1 con esta sustitución también muestran una disminución de la sensibilidad a la emtricitabina y a la lamivudina. Por lo tanto, puede presentarse resistencia cruzada entre estos fármacos en pacientes cuyos virus hospedan la sustitución K65R o K70E. Las cepas aisladas del VIH-1 de los pacientes (N = 20) cuyo VIH-1 expresó un promedio de tres sustituciones de la retrotranscriptasa asociadas a la zidovudina (M41L, D67N, K70R, L210W, T215Y/F o K219Q/E/N) mostraron una disminución de 3,1 veces de la sensibilidad al tenofovir. En los estudios 902 y 907 realizados en pacientes con tratamiento previo con antirretrovirales (VIREAD® + tratamiento de fondo estándar (TFE), en comparación con placebo + TFE) [véase Estudios clínicos], 14/304 (5 %) de los pacientes tratados con VIREAD® con fracaso virológico hasta la semana 96 tuvieron una disminución superior a 1,4 veces (mediana, 2,7 veces) de la sensibilidad al tenofovir. El análisis genotípico de las cepas aisladas iniciales y de fracaso mostró presentación de la sustitución K65R en el gen de la retrotranscriptasa del VIH-1. Se ha evaluado la respuesta virológica al tratamiento con VIREAD® con respecto al genotipo viral inicial (N = 222) en pacientes con tratamiento previo con antirretrovirales que participaron en los estudios 902 y 907. En estos estudios clínicos, el 94 % de los participantes evaluados tenían cepas aisladas del VIH-1 iniciales que expresaban al menos una sustitución para el inhibidor nucleosídico de la retrotranscriptasa (INRT). Las respuestas virológicas de los pacientes del subestudio de genotipos fueron similares a los resultados generales del estudio. Se realizaron varios análisis exploratorios para evaluar el efecto de sustituciones específicas y pautas mutacionales en los resultados virológicos. Debido al gran número de comparaciones potenciales, no se realizaron pruebas estadísticas. Se observaron varios grados de resistencia cruzada de VIREAD® a sustituciones asociadas con la resistencia a la zidovudina preexistentes (M41L, D67N, K70R, L210W, T215Y/F o K219Q/E/N) y parecían depender del tipo y el número de sustituciones específicas. Los pacientes tratados con VIREAD® cuyo VIH-1 expresó tres o más sustituciones asociadas con la resistencia a la zidovudina que incluyeron ya sea la sustitución M41L o la L210W de la retrotranscriptasa, presentaron respuestas reducidas al tratamiento con VIREAD®; sin embargo, estas respuestas fueron de todas maneras mejores en comparación con las respuestas al placebo. La presencia de las sustituciones D67N, K70R, T215Y/F o K219Q/E/N no pareció afectar las respuestas al tratamiento con VIREAD®. Los pacientes cuyo virus expresaba una sustitución L74V sin sustituciones asociadas con la resistencia a la zidovudina (N = 8) presentaron una disminución de la respuesta a VIREAD®. Se dispone de datos limitados en el caso de los pacientes cuyo virus expresaba una sustitución Y115F (N = 3), una sustitución Q151M (N = 2) o una inserción T69 (N = 4), todos los cuales presentaban una disminución de la respuesta. En los análisis definidos en el protocolo, la respuesta virológica a VIREAD® no disminuyó en los pacientes con infección por el VIH-1 que expresaron la sustitución M184V asociada con la resistencia a abacavir, emtricitabina y lamivudina. Las respuestas del ARN del VIH-1 en estos pacientes se mantuvieron hasta la semana 48. Análisis fenotípicos de los estudios 902 y 907: El análisis fenotípico del VIH-1 inicial de los pacientes que habían recibido tratamiento previo (N = 100) demostró una correlación entre la sensibilidad inicial a VIREAD® y la respuesta al tratamiento con VIREAD®. En la tabla 4 se resume la respuesta del ARN del VIH-1 según la sensibilidad inicial a VIREAD®.

Actividad contra el VHB: Actividad antiviral: La actividad antiviral del tenofovir contra el VHB se evaluó en la línea celular HepG2 2.2.15. Los valores de CE50 para el tenofovir variaron entre 0,14 y 1,5 mM, con valores de CC50 (50 % de la concentración citotóxica) superiores a 100 mM. No se observó actividad antagonista en los estudios de actividad antiviral en combinaciones de cultivos celulares realizados con el tenofovir y los inhibidores nucleosídicos de la retrotranscriptasa del VHB, entecavir, lamivudina, telbivudina o con el inhibidor nucleosídico de la retrotranscriptasa del VIH-1 emtricitabina. Resistencia: En los estudios 0102, 0103, 0106, 0108 y 0121, se evaluó anualmente la resistencia genotípica acumulativa a VIREAD® durante al menos 384 semanas con las secuencias apareadas de aminoácidos de la retrotranscriptasa del VHB en cepas aisladas obtenidas antes y durante el tratamiento, de pacientes que recibieron al menos 24 semanas de monoterapia con VIREAD® y que seguían presentando viremia con un nivel de ADN de VHB igual o superior a 400 copias/ml (69 UI/ml) al final de cada año del estudio (o en el momento de la suspensión de la monoterapia con VIREAD®), empleando un análisis según el tratamiento. En la población de pacientes sin tratamiento previo con nucleótidos de los estudios 0102 y 0103, los pacientes HBeAg+ presentaron una mayor carga viral inicial que los pacientes HBeAg-, y una proporción significativamente mayor de pacientes siguieron presentando viremia en el momento de la última medición durante la monoterapia con VIREAD® (15 % frente al 5%, respectivamente). Las cepas aisladas del VHB de estos pacientes que seguían presentando viremia mostraron sustituciones aparecidas durante el tratamiento (tabla 5); sin embargo, no se produjeron sustituciones específicas con una frecuencia suficiente para ser asociada con la resistencia a VIREAD® (análisis genotípicos o fenotípicos). Ver tabla 5.
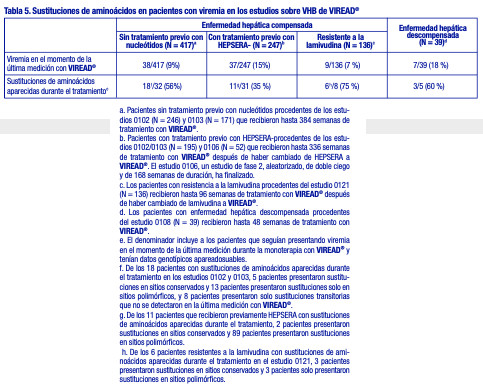
Resistencia cruzada: Se ha observado resistencia cruzada entre los inhibidores análogos nucleosídicos/nucleotídicos de la retrotranscriptasa del VHB. En estudios basados en células, las cepas del VHB que expresaron las sustituciones rtV173L, rtL180M y rtM204I/V asociadas con la resistencia a la lamivudina y la telbivudina mostraron una sensibilidad al tenofovir de 0,7 a 3,4 veces mayor en comparación con el virus natural. Las sustituciones dobles rtL180M y rtM204I/V redujeron 3,4 veces la sensibilidad al tenofovir. Las cepas del VHB que expresaron las sustituciones rtL180M, rtT184G, rtS202G/I, rtM204V y rtM250V asociadas con la resistencia al entecavir mostraron una sensibilidad al tenofovir de 0,6 a 6,9 veces mayor en comparación con el virus natural. Las cepas del VHB que expresaron las sustituciones rtA181V y/o rtN236T asociadas con la resistencia al adefovir redujeron de 2,9 a 10 veces la sensibilidad al tenofovir en comparación con el virus natural. Las cepas que presentaban la sustitución rtA181T mostraron cambios en la sensibilidad al tenofovir de 0,9 a 1,5 veces mayor en comparación con el virus natural. En los estudios 0102, 0103, 0106, 0108 y 0121, 152 pacientes que iniciaron el tratamiento con VIREAD® albergaron VHB con sustituciones de resistencia conocidas a los inhibidores análogos nucleos(t)ídicos de la retrotranscriptasa del VHB: 14 con sustituciones asociadas con la resistencia al adefovir (rtA181T/V y/o rtN236T), 135 con sustituciones asociadas con la resistencia a la lamivudina (rtM204I/V) y 3 con sustituciones resistentes tanto al adefovir como a la lamivudina. Luego de hasta 384 semanas de tratamiento con VIREAD®, 10 de los 14 pacientes con VHB resistente al adefovir, 124 de los 135 pacientes con VHB resistente a la lamivudina y 2 de los 3 pacientes con VHB resistente tanto al adefovir como a la lamivudina lograron y mantuvieron la supresión virológica (ADN de VHB inferior a 400 copias/ml [69 UI/ml]). Tres de los 5 pacientes cuyo virus albergaba las dos sustituciones rtA181T/V y rtN236T siguieron presentando viremia.
Indicaciones.
Infección por VIH-1: VIREAD® está indicado en asociación con otros antirretrovirales para el tratamiento de la infección causada por el VIH-1 en adultos y pacientes pediátricos de 12 años o más. Los siguientes aspectos deberán tenerse en cuenta al iniciar la terapia con VIREAD® para el tratamiento de la infección por el VIH-1: VIREAD® no se debe administrar concomitantemente con otros medicamentos que contengan tenofovir disoproxil fumarato o tenofovir alafenamida [véase Advertencias]. Hepatitis B crónica: VIREAD® está indicado para el tratamiento de la hepatitis B crónica en adultos y pacientes pediátricos de 12 años o más. Los siguientes aspectos deberán tenerse en cuenta al iniciar la terapia con VIREAD® para el tratamiento de la infección crónica por hepatitis B: Esta indicación en adultos se basa en los datos de seguridad y eficacia obtenidos del tratamiento en pacientes que no habían recibido previamente nucleósidos y en pacientes que habían recibido tratamiento previo con resistencia documentada a la lamivudina. Los pacientes eran adultos que tenían hepatitis B crónica HBeAg positiva y HBeAg negativa con enfermedad hepática compensada [véase Eficacia clínica en adultos con hepatitis B crónica]. VIREAD® fue evaluado en una cantidad limitada de pacientes con hepatitis B crónica y enfermedad hepática descompensada [véase Reacciones adversas, Eficacia clínica en adultos con hepatitis B crónica]. En los ensayos clínicos, la cantidad de pacientes que tenían sustituciones asociadas con resistencia al adefovir al inicio fue demasiado reducida para obtener conclusiones sobre la eficacia [véase Microbiología, Eficacia clínica en adultos con hepatitis B crónica].
Dosificación.
Dosis recomendada en adultos y pacientes pediátricos de 12 años o más (35kg o más): Para el tratamiento del VIH-1 o la hepatitis B crónica: La dosis es un comprimido de 300 mg de VIREAD® una vez al día, administrado por vía oral con o sin alimentos. Para el tratamiento de la hepatitis B crónica, se desconoce la duración óptima del tratamiento. Ajuste de la dosis en caso de disfunción renal en adultos: Se produjo un aumento significativo de las exposiciones al fármaco cuando se administró VIREAD® a pacientes con disfunción renal de moderada a grave [véase Farmacología clínica]. Por lo tanto, el intervalo de dosificación de VIREAD® debe ajustarse en los pacientes con una depuración de creatinina inicial inferior a 50 ml/min, aplicando las recomendaciones de la tabla 6. Las recomendaciones de ajustes en el intervalo de las dosis se basan en modelos de datos farmacocinéticos de dosis única en los pacientes no infectados por el VIH o por el VHB con diferentes grados de disfunción renal, incluso enfermedad renal terminal que precisa hemodiálisis. No se han evaluado clínicamente la seguridad ni la eficacia de estas recomendaciones de ajustes en el intervalo de las dosis en los pacientes con disfunción renal moderada o grave; por lo tanto, en estos pacientes se deben controlar rigurosamente la respuesta clínica al tratamiento y la función renal [véase Advertencias]. No es necesario ajustar la dosis en los pacientes con insuficiencia renal leve (depuración de creatinina de 50 a 80 ml/min). Debe vigilarse periódicamente la depuración de creatinina estimada, el fósforo sérico, la glucosa en orina y la proteinuria en los pacientes con disfunción renal leve [véase Advertencias].

No se han evaluado las propiedades farmacocinéticas del tenofovir en los pacientes que no estén en hemodiálisis, con un valor de depuración de creatinina inferior a 10 ml/min; por lo tanto, no se dispone de una dosis recomendada para estos pacientes. No hay datos disponibles que permitan hacer recomendaciones sobre la dosis a utilizar en pacientes pediátricos de 12 años o más con disfunción renal. Formas farmacéuticas y concentraciones: VIREAD® se comercializa en forma de comprimidos. Cada comprimido contiene 300 mg de tenofovir disoproxil fumarato, lo que es equivalente a 245 mg de disoproxilo de tenofovir. Los comprimidos son de forma de almendra, de color azul claro, recubiertos con película, y llevan la inscripción "GILEAD" y "4331" en un lado y "300" en el otro lado.
Contraindicaciones.
Hipersensibilidad al principio activo o a cualquiera de los excipientes.
Reacciones adversas.
En otros apartados de la ficha técnica se tratan las siguientes reacciones adversas: Exacerbación aguda y grave de la hepatitis [véase Advertencias]. Nueva aparición o empeoramiento de la disfunción renal [véase Advertencias]. Acidosis láctica/Severa hepatomegalia con esteatosis [Consulte Advertencias]. Efectos óseos [véase Advertencias]. Síndrome de reconstitución inmunológica [véase Advertencias]. Reacciones adversas de la experiencia en ensayos clínicos: Debido a que los ensayos clínicos se realizan en condiciones muy variadas, las tasas de reacciones adversas observadas en los ensayos clínicos de un fármaco no pueden compararse directamente con las tasas en los ensayos clínicos de otro fármaco y puede que no reflejen las tasas observadas en la práctica. Ensayos clínicos en pacientes adultos con infección por VIH-1: Se ha tratado a más de 12 000 pacientes con VIREAD® solo o en asociación con otros antirretrovirales, durante períodos de 28 días hasta 215 semanas, en ensayos clínicos y en programas de acceso ampliado. Un total de 1544 pacientes han recibido VIREAD® en dosis de 300 mg una vez al día en los ensayos clínicos; más de 11 000 pacientes han recibido VIREAD® en programas de acceso ampliado. Las reacciones adversas más frecuentes (incidencia superior o igual al 10 %, grados 2 4) identificadas a partir de cualquiera de los tres grandes ensayos clínicos controlados son erupción cutánea, diarrea, cefalea, dolor, depresión, astenia y náuseas. Pacientes sin tratamiento previo con antirretrovirales: Estudio 903. Reacciones adversas aparecidas con el tratamiento: Las reacciones adversas más frecuentes que se observaron en un estudio de comparación, controlado, y con doble enmascaramiento ("doble ciego"), en el que 600 pacientes sin tratamiento previo con antirretrovirales recibieron VIREAD® (N = 299) o estavudina (N = 301) en asociación con lamivudina y efavirenz durante 144 semanas (Estudio 903) fueron mareos y reacciones digestivas de leves a moderadas. Las reacciones adversas leves (grado 1) fueron frecuentes, con una incidencia similar en ambos grupos de tratamiento, y consistieron en mareos, diarrea y náuseas. En la tabla 12 se resumen algunas de las reacciones adversas de moderadas a graves que aparecieron con el tratamiento.
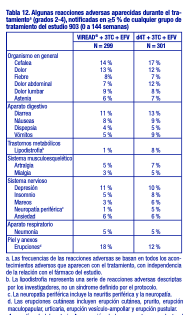
Anomalías de laboratorio: A excepción de los aumentos en colesterol y triglicéridos en ayunas que fueron más frecuentes en el grupo tratado con estavudina (40 % y 9 %) en comparación con el grupo tratado con VIREAD® (19 % y 1 %), respectivamente, las anomalías de laboratorio observadas en este estudio ocurrieron con frecuencias similares en los grupos tratados con VIREAD® y con estavudina. En la tabla 13 se presenta un resumen de las anomalías de laboratorio de grados 3-4.
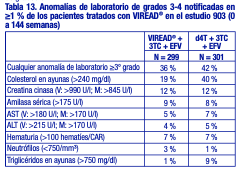
Estudio 934. Reacciones adversas aparecidas con el tratamiento: En el estudio 934, se administró a 511 pacientes sin tratamiento previo con antirretrovirales VIREAD® + EMTRIVA® (emtricitabina) en combinación con efavirenz (N = 257), o zidovudina y lamivudina en combinación con efavirenz (N = 254). Las reacciones adversas observadas en este estudio fueron en general coherentes con las observadas en estudios anteriores en los pacientes con o sin tratamiento previo con antirretrovirales (tabla 14). Cambios en la densidad mineral ósea: En los pacientes adultos infectados por el VIH-1 en el estudio 903, se observó una disminución en el porcentaje medio significativamente superior, con respecto al valor inicial, de la DMO de la columna lumbar en los pacientes tratados con VIREAD® + lamivudina + efavirenz (-2,2 % ± 3,9), en comparación con los pacientes tratados con estavudina + lamivudina + efavirenz (-1,0 % ± 4,6) durante 144 semanas. Los cambios en la DMO de la cadera fueron similares en los dos grupos de tratamiento (-2,8 % ± 3,5 en el grupo tratado con VIREAD® en comparación con el -2,4 % ± 4,5 en el grupo que recibió estavudina). En ambos grupos, la mayoría de la reducción de la DMO se produjo durante las primeras 24 a 48 semanas del estudio y se mantuvo durante 144 semanas. El 28 % de los pacientes tratados con VIREAD®, en comparación con el 21 % de los que recibieron estavudina, perdieron por lo menos el 5 % de la DMO en la columna o el 7 % de la DMO en la cadera. Se notificaron fracturas clínicamente relevantes (excepto de los dedos de los pies y las manos) en cuatro pacientes del grupo que recibió VIREAD® y en seis pacientes del grupo tratado con estavudina. Además, hubo aumentos significativos en los marcadores bioquímicos del metabolismo óseo (fosfatasa alcalina específica del tejido óseo sérica, osteocalcina sérica, telopéptido C sérico y telopéptido N urinario) y mayores concentraciones séricas de la hormona paratiroidea y de la 1,25-vitamina D en el grupo tratado con VIREAD®, en comparación con el grupo que recibió estavudina. Sin embargo, a excepción de la fosfatasa alcalina específica del tejido óseo, estos cambios dieron como resultado valores que se mantuvieron dentro de los límites de la normalidad [véase Advertencias].
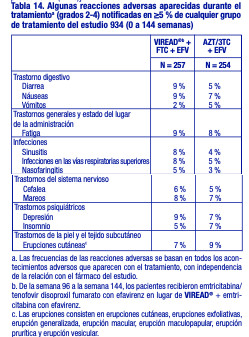
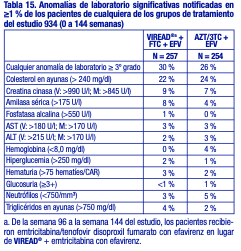
Anomalías de laboratorio: Las anomalías de laboratorio observadas en este estudio fueron en general coherentes con las observadas en estudios anteriores (tabla 15).
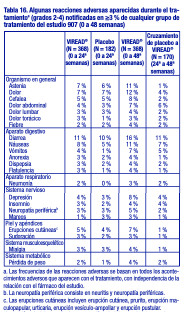
Pacientes con tratamiento previo con antirretrovirales: Reacciones adversas aparecidas con el tratamiento: Las reacciones adversas observadas en los pacientes con tratamiento previo fueron generalmente coherentes con aquellas observadas en los pacientes sin tratamiento previo, entre ellas, reacciones digestivas de leves a moderadas, como náuseas, diarrea, vómitos y flatulencia. Menos del 1 % de los pacientes interrumpieron su participación en los estudios clínicos debido a reacciones adversas digestivas (estudio 907). En la tabla 16 se presenta un resumen de las reacciones adversas de moderadas a graves, aparecidas con el tratamiento, que se presentaron durante las primeras 48 semanas del estudio 907.
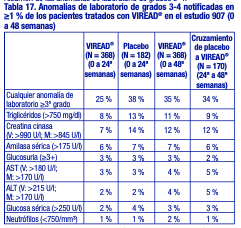
Anomalías de laboratorio: Las anomalías de laboratorio observadas en este estudio se presentaron con frecuencias similares en los grupos tratados con placebo y con VIREAD®. En la tabla 17 se presenta un resumen de las anomalías de laboratorio de grados 3-4.
Ensayos clínicos en pacientes pediátricos de 12 años o más con infección por VIH-1: La evaluación de las reacciones adversas se basa en un estudio aleatorizado (estudio 321) en 87 pacientes pediátricos (12 a menos de 18 años) infectados por el VIH-1 que recibieron tratamiento con VIREAD® (N = 45) o placebo (N = 42) en combinación con otros antirretrovirales durante 48 semanas. Las reacciones adversas observadas en los adolescentes que recibieron tratamiento con VIREAD® fueron coherentes con las observadas en los ensayos clínicos
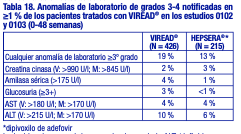
con adultos. Cambios en la densidad mineral ósea: Los estudios clínicos en niños y adolescentes infectados por el VIH-1 evaluaron los cambios en la DMO. En el estudio 321 (12 a menos de 18 años), la tasa media de ganancia de DMO en la semana 48 fue menor en el grupo tratado con VIREAD® que en el grupo de placebo. Seis pacientes tratados con VIREAD® y un paciente tratado con placebo presentaron una pérdida significativa (superior al 4%) de la DMO de la columna lumbar en la semana 48. Entre los 28 pacientes que recibieron VIREAD® durante 96 semanas, los cambios de las puntuaciones Z con respecto al valor inicial fueron de -0,341 para la columna lumbar y de -0,458 para el cuerpo completo. El crecimiento del esqueleto (altura) pareció no verse afectado [véase Advertencias]. Ensayos clínicos en pacientes adultos con hepatitis B crónica y enfermedad hepática compensada: Reacciones adversas aparecidas con el tratamiento: En los estudios clínicos controlados en los que participaron 641 pacientes con hepatitis B crónica (0102 y 0103), una mayor cantidad de pacientes tratados con VIREAD® durante el período de 48 semanas con doble ciego presentó náuseas: el 9 % con VIREAD® frente al 2 % con HEPSERA® (dipivoxilo de adefovir). Otras reacciones adversas aparecidas con el tratamiento que se informaron en más del 5 % de los pacientes tratados con VIREAD® incluyen: dolor abdominal, diarrea, dolor de cabeza, mareos, fatiga, nasofaringitis, dolor de espalda y erupciones cutáneas. Durante la fase abierta del tratamiento con VIREAD® (semanas 48 a 384) en los estudios 0102 y 0103, el 2% de los pacientes (13/585) experimentó un aumento confirmado en la creatinina sérica de 0,5 mg/dl con respecto al valor inicial. No se observaron cambios significativos en el perfil de tolerabilidad tras continuar el tratamiento con VIREAD® durante un máximo de 240 semanas. Anomalías de laboratorio: En la tabla 18, se presenta un resumen de las anomalías de laboratorio de grados 3-4 hasta la semana 48. Las anomalías de laboratorio de grados 3-4 fueron similares en los pacientes que continuaron el tratamiento con VIREAD® durante un máximo de 384 semanas en estos estudios.
La incidencia general de exacerbaciones de la ALT (definidas como ALT sérica superior a 2 × valor inicial y superior a 10 × LSN, con o sin síntomas asociados) durante el tratamiento fue similar entre VIREAD® (2,6 %) y HEPSERA® (dipivoxilo de adefovir) (2 %). En general, las exacerbaciones de la ALT se produjeron dentro de las primeras 4 a 8 semanas de tratamiento y estuvieron acompañadas de disminuciones en los niveles de ADN de VHB. Ninguno de los pacientes presentó indicios de descompensación. Las exacerbaciones de la ALT se resolvieron habitualmente en el plazo de 4 a 8 semanas sin modificar la medicación del estudio. Las reacciones adversas observadas en los pacientes con hepatitis crónica B y resistencia a la lamivudina que habían recibido tratamiento con VIREAD® fueron coherentes con las observadas en otros estudios de hepatitis B realizados en adultos. Estudios clínicos en pacientes adultos con hepatitis B crónica y enfermedad hepática descompensada: En un pequeño estudio aleatorizado, doble ciego, con control activo (0108), se administró tratamiento con VIREAD® u otros fármacos antivirales durante un máximo de 48 semanas a pacientes con hepatitis B crónica (HBC) y enfermedad hepática descompensada [véase Estudios clínicos]. Entre los 45 pacientes que recibieron VIREAD®, las reacciones adversas de cualquier nivel de gravedad aparecidas con el tratamiento que se informaron con mayor frecuencia fueron dolor abdominal (22 %), náuseas (20 %), insomnio (18 %), prurito (16 %), vómitos (13 %), mareos (13 %) y pirexia (11 %). Hasta la semana 48 del estudio, dos (4 %) de los 45 pacientes murieron debido al avance de la enfermedad hepática. Tres (7 %) de los 45 pacientes suspendieron el tratamiento debido a una reacción adversa. Cuatro (9 %) de los 45 pacientes presentaron un aumento confirmado de 0,5 mg/dl en la creatinina sérica (hasta la semana 48, 1 paciente también presentó un nivel confirmado de fósforo sérico inferior a 2 mg/dl). Tres de estos pacientes (cada uno de ellos tenía una puntuación superior o igual a 10 en la escala de Child-Pugh y una puntuación superior o igual a 14 en el modelo para enfermedad hepática terminal MELD en el momento del ingreso) presentaron insuficiencia renal. Debido a que tanto VIREAD® como la enfermedad hepática descompensada pueden afectar a la función renal, es difícil de precisar en qué medida VIREAD® contribuyó a la disfunción renal en esta población. Uno de los 45 pacientes presentó una exacerbación hepática mientras estaba en tratamiento durante el estudio de 48 semanas. Estudios clínicos en pacientes pediátricos de 12 años o más con hepatitis B crónica: La evaluación de las reacciones adversas se basa en un estudio aleatorizado (estudio GS-US-174-0115) en 106 pacientes pediátricos (12 a menos de 18 años de edad) infectados por hepatitis B crónica que recibieron tratamiento con VIREAD® (N = 52) o placebo (N = 54) durante 72 semanas. Las reacciones adversas observadas en los pacientes pediátricos que recibieron tratamiento con VIREAD® fueron coherentes con las observadas en los estudios clínicos de VIREAD® en adultos. En este estudio, tanto el grupo de VIREAD® como el grupo de placebo experimentaron un incremento global en la DMO media de la columna lumbar durante 72 semanas, tal como se esperaba en la población adolescente. Las ganancias de DMO desde el inicio hasta la semana 72 en la DMO de la columna lumbar y el cuerpo completo de los pacientes tratados con VIREAD® (+5 % y +3 %, respectivamente) fueron inferiores a las ganancias de DMO observadas en los pacientes tratados con placebo (+8 % y +5 %, respectivamente). Tres pacientes del grupo de VIREAD® y dos pacientes del grupo de placebo presentaron pérdida significativa (superior al 4 %) de la DMO de la columna lumbar en la semana 72. Al inicio, las puntuaciones Z de la DMO media en los pacientes aleatorizados a VIREAD® fueron de -0,43 para la columna lumbar y de -0,20 para el cuerpo completo, y las puntuaciones Z de la DMO media en los pacientes aleatorizados a placebo fueron de -0,28 para la columna lumbar y de -0,26 para el cuerpo completo. En los pacientes que recibieron VIREAD® durante 72 semanas, el cambio medio en la puntuación Z de la DMO fue de -0,05 para la columna lumbar y de -0,15 para el cuerpo completo, en comparación con un +0,07 y +0,06, respectivamente, en los pacientes que recibieron placebo. Como se observó en los estudios pediátricos con pacientes infectados por el VIH, el crecimiento del esqueleto (altura) pareció no verse afectado [véase Advertencias]. Experiencia durante la comercialización: Se han identificado las siguientes reacciones adversas durante el uso posterior a la aprobación de VIREAD®. Debido a que las reacciones posteriores a la comercialización se notifican voluntariamente a partir de una población de tamaño indeterminado, no siempre es posible calcular de manera fiable su frecuencia ni establecer una relación causal con la exposición al fármaco. Trastornos del sistema inmunitario: Reacción alérgica (incluso angioedema). Trastornos de la nutrición y el metabolismo: Acidosis láctica, hipopotasemia, hipofosfatemia. Trastornos respiratorios, torácicos y mediastínicos: Disnea. Trastornos digestivos: Pancreatitis, aumento de la amilasa, dolor abdominal. Trastornos hepatobiliares: Esteatosis hepática, hepatitis, aumento de las enzimas hepáticas (con mayor frecuencia, de la AST, la ALT, la gamma GT). Trastornos de la piel y el tejido subcutáneo: Erupción cutánea. Trastornos musculoesqueléticos y del tejido conjuntivo: Rabdomiólisis, osteomalacia (manifestada como dolor óseo y que puede contribuir a las fracturas), debilidad muscular, miopatía. Trastornos renales y urinarios: Insuficiencia renal aguda, insuficiencia renal, necrosis tubular aguda, síndrome de Fanconi, tubulopatía renal proximal, nefritis intersticial (incluso casos agudos), diabetes insípida nefrógena, disfunción renal, aumento de la creatinina, proteinuria, poliuria. Trastornos generales y alteraciones en el lugar de la administración: Astenia. Las siguientes reacciones adversas, enumeradas bajo los encabezados de los sistemas corporales anteriores, pueden producirse a consecuencia de la tubulopatía renal proximal: rabdomiólisis, osteomalacia, hipopotasemia, debilidad muscular, miopatía, hipofosfatemia.
Advertencias.
Generales: Exacerbación de la hepatitis después de la suspensión del tratamiento: La suspensión del tratamiento contra el VHB, incluido VIREAD®, puede estar asociada con exacerbaciones agudas y graves de la hepatitis. Se debe controlar rigurosamente a los pacientes infectados por el VHB que suspendan la administración de VIREAD®, con seguimiento clínico y de laboratorio durante por lo menos varios meses después de la suspensión del tratamiento. Si fuese conveniente, puede estar justificada la reanudación del tratamiento contra la hepatitis B. Nueva aparición o empeoramiento de la disfunción renal: El tenofovir se elimina principalmente por los riñones. Se han notificado casos de trastornos renales, entre ellos casos de insuficiencia renal aguda y síndrome de Fanconi (lesión tubular renal con hipofosfatemia grave), asociados con el uso de VIREAD® [véase Reacciones adversas]. Se recomienda evaluar la depuración de creatinina estimada en todos los pacientes antes de iniciar la terapia y según se requiera clínicamente durante el tratamiento con VIREAD®. En los pacientes con riesgo de disfunción renal, incluso los pacientes que han sufrido anteriormente alteraciones renales mientras recibían dipivoxilo de adefovir, se recomienda evaluar la depuración de creatinina estimada, el fósforo sérico, la glucosa en orina y la proteinuria antes de iniciar el tratamiento con VIREAD®, y de forma periódica durante el tratamiento con VIREAD®. Se recomienda el ajuste del intervalo de dosificación de VIREAD® y la vigilancia rigurosa de la función renal en todos los pacientes con una depuración de creatinina inferior a 50 ml/min [véase Dosificación]. No hay información sobre la inocuidad ni la eficacia en los pacientes con disfunción renal que hayan recibido VIREAD® siguiendo estas pautas de dosificación, por lo que el beneficio potencial del tratamiento con VIREAD® debe evaluarse en relación con el posible riesgo de la toxicidad renal. Se debe evitar el uso de VIREAD® con la utilización reciente o concomitante de un medicamento nefrotóxico (p. ej., múltiples antinflamatorios no esteroideos [AINE] o a dosis altas) [véase Interacciones medicamentosas]. Se han notificado casos de disfunción renal grave tras la administración de múltiples AINE o a dosis altas en pacientes infectados por el VIH con factores de riesgo de disfunción renal que parecían estables con tenofovir DF. Algunos pacientes tuvieron que ser internados y recibir un tratamiento renal sustitutivo. En caso de ser necesario, deben considerarse alternativas terapéuticas a los AINE en los pacientes con riesgo de disfunción renal. El dolor óseo persistente o empeoramiento del mismo, el dolor en las extremidades, las fracturas y/o el dolor o la debilidad muscular pueden ser manifestaciones de tubulopatía renal proximal y requerirán una evaluación de la función renal de los pacientes en riesgo. Acidosis láctica/Hepatomegalia severa con esteatosis: Se han informado casos de acidosis láctica y hepatomegalia grave con esteatosis, incluso casos mortales, con el uso de análogos de nucleosidos, incluido el tenofovir DF, solos o en combinación con otros antirretrovirales. El tratamiento con VIREAD deberá ser suspendido en caso de que algún paciente desarrolle manifestaciones clínicas y/o parámetros de laboratorio sugestivos de acidosis láctica o hepatotoxicidad pronunciada (que pueden incluir hepatomegalia y esteatosis incluso en ausencia de elevación de transaminasas). Administración concomitante con otros productos: VIREAD® no debe utilizarse junto con los productos de asociación en dosis fijas que contengan tenofovir DF entre sus componentes. VIREAD® no deberá administrarse en asociación con dipivoxilo de adefovir [véase Interacciones medicamentosas]. Pacientes con infección concomitante por el VIH-1 y el VHB: Debido al riesgo de desarrollar resistencia al VIH-1, VIREAD® debe utilizarse únicamente en pacientes infectados concomitantemente por el VIH-1 y el VHB como parte de un régimen adecuado antirretroviral combinado. Deben ofrecerse pruebas de anticuerpos contra el VIH-1 a todos los pacientes infectados por el VHB antes de iniciar el tratamiento con VIREAD®. También se recomienda que se hagan análisis en todos los pacientes infectados por el VIH-1 para detectar la presencia de hepatitis B crónica antes de iniciar el tratamiento con VIREAD®. Efectos óseos: Densidad mineral ósea: En los estudios clínicos con adultos infectados por el VIH-1, VIREAD® se asoció a disminuciones ligeramente superiores de la densidad mineral ósea (DMO) y a aumentos en los marcadores bioquímicos del metabolismo óseo, lo que sugiere un mayor recambio óseo frente a los comparadores. Las concentraciones séricas de la hormona paratiroidea y de 1,25-vitamina D también fueron más altas en los pacientes tratados con VIREAD® [véase Reacciones adversas]. Se realizaron estudios clínicos para evaluar VIREAD® en pacientes pediátricos y adolescentes. En circunstancias normales, la DMO aumenta rápidamente en los pacientes pediátricos. En los pacientes de 2 a menos de 18 años de edad infectados por el VIH-1, los efectos óseos fueron similares a los observados en los pacientes adultos, lo que sugiere un mayor recambio óseo. La ganancia total de DMO corporal fue menor en el grupo de pacientes pediátricos infectados por el VIH-1 tratados con VIREAD® que en los grupos de control. Se observaron tendencias similares en los pacientes adolescentes de 12 a menos de 18 años de edad con hepatitis B crónica. En todos los estudios pediátricos, el crecimiento del esqueleto (estatura) no pareció resultar afectado. [Véase Reacciones adversas]. Se desconocen los efectos que los cambios asociados con VIREAD® en la DMO y en los marcadores bioquímicos pueden tener en la salud ósea a largo plazo y en el riesgo futuro de fracturas. Se debe plantear la evaluación de la DMO en los adultos y pacientes pediátricos de 12 años o más que tienen antecedentes de fracturas óseas patológicas u otros factores de riesgo de osteoporosis o pérdida ósea. Si bien no se ha estudiado el efecto de los suplementos de calcio y vitamina D, dichos suplementos pueden ser beneficiosos para todos los pacientes. Se debe obtener asesoramiento adecuado si se sospecha de la presencia de anomalías óseas. Defectos de mineralización: Se han notificado casos de osteomalacia asociada a tubulopatía renal proximal, que se manifiesta como dolor óseo o dolor en las extremidades y que puede contribuir a las fracturas, en relación con el uso de tenofovir DF [véase Reacciones adversas]. Asimismo, se han notificado artralgias y dolor o debilidad muscular en casos de tubulopatía renal proximal. Deben tenerse en cuenta la hipofosfatemia y la osteomalacia secundaria a tubulopatía renal proximal en los pacientes con riesgo de disfunción renal que presentan síntomas musculares u óseos persistentes o un empeoramiento de los mismos mientras reciben tratamiento con fármacos que contienen tenofovir DF [véase Advertencias]. Síndrome de reconstitución inmunológica: Se ha informado la aparición del síndrome de reconstitución inmunológica en los pacientes infectados por el VIH y que reciben tratamiento antirretroviral combinado, incluyendo VIREAD®. Durante la fase inicial del tratamiento antirretroviral asociado, los pacientes cuyo sistema inmunitario responde pueden presentar una respuesta inflamatoria ante infecciones oportunistas residuales o indolentes (por ejemplo, infección por Mycobacterium avium, citomegalovirus, neumonía por Pneumocystis jirovecii o tuberculosis), que pueden requerir evaluación y tratamiento adicionales. También se han notificado casos de trastornos autoinmunitarios (como enfermedad de Graves, polimiositis y síndrome de Guillain Barré), que se produjeron en el contexto de reconstitución inmunitaria; sin embargo, el momento de la aparición de estos trastornos es más variable, y pueden aparecer muchos meses después del inicio del tratamiento. Fracaso virológico temprano: Los estudios clínicos en pacientes infectados por el VIH han demostrado que algunas pautas que contienen solamente tres inhibidores nucleosídicos de la retrotranscriptasa (INRT) son generalmente menos eficaces que las pautas de tres medicamentos que contienen dos INRT en combinación con algún inhibidor no nucleosídico de la retrotranscriptasa o un inhibidor de la proteasa del VIH-1. En particular, se han notificado fracasos virológicos tempranos y tasas elevadas de sustituciones de resistencia. Por lo tanto, las pautas de tres nucleósidos deberán utilizarse con precaución. Se debe controlar rigurosamente a los pacientes que estén utilizando solamente una pauta con tres nucleósidos y se debe plantear la modificación de su tratamiento.
Interacciones.
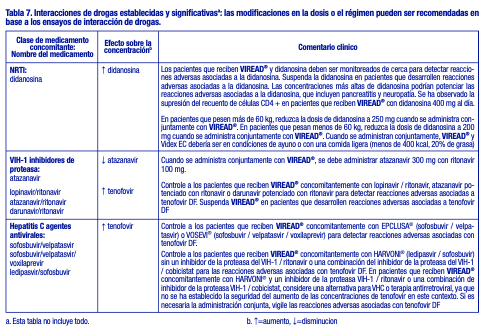
Fármacos que afectan a la función renal: El tenofovir se elimina principalmente por los riñones [véase Farmacología clínica], la administración concomitante de VIREAD® con fármacos que son eliminados por secreción tubular activa puede aumentar las concentraciones del tenofovir y/o las de otros fármacos coadministrados. Algunos ejemplos son, entre otros, aciclovir, cidofovir, ganciclovir, valaciclovir, valganciclovir, aminoglucósidos (p. ej., gentamicina) y múltiples AINE o a dosis altas [véase Advertencias]. Los medicamentos que disminuyen la función renal pueden aumentar las concentraciones de tenofovir. En el tratamiento de la hepatitis B crónica, VIREAD® no debe administrarse en combinación con dipivoxilo de adefovir. Interacciones establecidas y significativas: La Tabla 7 proporciona una lista de interacciones farmacológicas establecidas o clínicamente significativas. Las interacciones farmacológicas descritas se basan en estudios realizados con tenofovir DF [consulte Farmacología clínica]. Ver Tabla 7.
Toxicología no clínica: Carcinogénesis, mutagénesis, deterioro de la fertilidad: Carcinogénesis: Se realizaron estudios a largo plazo de la carcinogénesis oral del tenofovir DF en ratas y ratones, con exposiciones de hasta aproximadamente 16 veces (ratones) y 5 veces (ratas) más que las observadas en los seres humanos con la dosis terapéutica para la infección por el VIH-1. Con la dosis alta en ratones hembra, aumentaron los adenomas hepáticos con exposiciones 16 veces superiores a las de los seres humanos. En las ratas, el estudio arrojó resultados carcinogénicos negativos con exposiciones de hasta 5 veces las observadas en los seres humanos con la dosis terapéutica. Mutagénesis: El tenofovir DF fue mutagénico en la prueba de linfoma en ratones in vitro y dio un resultado negativo en la prueba de mutagénesis bacteriana in vitro (prueba de Ames). En un estudio de micronúcleos en ratones in vivo, el tenofovir DF resultó negativo cuando se administró a ratones macho. Deterioro de la fertilidad: No hubo efectos en la fertilidad, la capacidad de apareamiento ni el desarrollo embrionario temprano cuando se administró el tenofovir DF a ratas macho, a una dosis equivalente a 10 veces la dosis en los seres humanos de acuerdo con las comparaciones de la superficie corporal, en los 28 días previos al apareamiento, y a ratas hembra, durante 15 días antes del apareamiento hasta el séptimo día de gestación. Sin embargo, hubo una alteración del ciclo estral en las ratas hembra. Toxicología y/o farmacología en animales: El tenofovir y el tenofovir DF administrados a ratas, perros y monos en estudios toxicológicos con exposiciones (según el AUC) superiores o iguales a seis veces las observadas en los seres humanos ocasionaron toxicidad ósea. En los monos, la toxicidad ósea se diagnosticó como osteomalacia. La osteomalacia observada en los monos parecía ser reversible al reducir la dosis o suspender el tenofovir. En las ratas y los perros, la toxicidad ósea se manifestó en forma de disminución de la densidad mineral ósea. Se desconocen los mecanismos subyacentes de la toxicidad ósea. Se obtuvieron indicios de toxicidad renal en cuatro especies animales. En estos animales se observaron aumentos en la creatinina sérica, nitrógeno ureico en sangre, glucosuria, proteinuria, fosfaturia y/o calciuria y disminución del fosfato sérico en diferentes grados. Estas toxicidades se evidenciaron en exposiciones (según el AUC) de 2 a 20 veces superiores a las observadas en los seres humanos. Se desconoce la relación de las anomalías renales, en especial de la fosfaturia, con la toxicidad ósea. 7.4 Uso en poblaciones específicas Embarazo: Embarazo categoría B: No existen estudios adecuados y bien controlados en mujeres embarazadas. Debido a que los estudios de reproducción en animales no siempre predicen la respuesta en los seres humanos, sólo se debe administrar VIREAD® durante el embarazo si es estrictamente necesario. Registro de embarazos con antirretrovirales: A fin de monitorear los resultados fetales de las mujeres embarazadas expuestas a VIREAD®, se ha establecido un registro de embarazos con antirretrovirales (Antiretroviral Pregnancy Registry, APR). Se recomienda a los médicos que registren a las pacientes que se queden embarazadas en el sitio web: http://www.apregistry.com, o que lo comuniquen llamando al +54 11 4858 9000 (extensión 229) o enviando un correo electrónico a farmacovigilancia@gador.com.ar. Resumen de los riesgos: Datos en animales: Los estudios sobre reproducción se realizaron en ratas y conejos con dosis de hasta 14 y 19 veces las dosis en los seres humanos, de acuerdo con las comparaciones de la superficie corporal, y no revelaron ningún indicio de trastorno de la fertilidad o daño al feto debido al tenofovir. Lactancia: Se recomienda que las madres infectadas por el VIH-1 no amamanten a sus hijos, a fin de evitar el riesgo de transmisión posnatal del VIH-1. Las muestras de leche materna obtenidas a partir de cinco madres infectadas por el VIH-1 durante la primera semana postparto muestran que tenofovir se excreta por la leche materna. Se desconoce el impacto de dicha exposición en los lactantes. Debido a la posibilidad tanto de transmisión del VIH-1 como de reacciones adversas graves en los lactantes, se debe indicar a las madres que no amamanten si reciben VIREAD®. Uso pediátrico: Pacientes pediátricos de 12 años o más con infección por el VIH-1: La seguridad de VIREAD® en pacientes pediátricos de 12 a menos de 18 años de edad está respaldada por los datos de un estudio aleatorizado en el que se administró VIREAD® a pacientes infectados por el VIH-1 que habían recibido tratamiento previo con antirretrovirales. En este estudio, el perfil farmacocinético de VIREAD® fue similar al que había demostrado ser seguro y eficaz en los ensayos clínicos con adultos [véase Farmacología clínica]. En el estudio 321, 87 pacientes de 12 a menos de 18 años de edad que habían recibido tratamiento previo con antirretrovirales fueron tratados con VIREAD® (N = 45) o placebo (N = 42) junto con un régimen de fondo optimizado durante 48 semanas. El recuento medio inicial de linfocitos CD4 fue de 374 linfocitos/mm3 y el valor basal medio de ARN del VIH-1 en plasma fue de 4,6 log10 copias/ml. Al inicio, el 90 % de los pacientes presentaban sustituciones asociadas con la resistencia a los INRT en sus cepas aisladas de VIH-1. En general, el ensayo no mostró ninguna diferencia en la respuesta virológica entre los grupos de tratamiento de VIREAD® y de placebo. Los análisis de los subgrupos indican que la ausencia de una diferencia en la respuesta virológica podría atribuirse a desequilibrios entre los grupos de tratamiento en cuanto a la sensibilidad viral basal a VIREAD® y al régimen de fondo optimizado. Aunque los cambios en el ARN del VIH-1 en estos pacientes con gran cantidad de tratamientos antirretrovirales previos fueron menores a los esperados, la comparabilidad de los datos farmacocinéticos y de seguridad con respecto a los datos observados en los adultos respalda el uso de VIREAD® en pacientes pediátricos de 12 años o más con un peso corporal superior o igual a 35 kg, y cuyas cepas aisladas de VIH-1 se espera que sean sensibles a VIREAD® [véanse Advertencias, Reacciones adversas y Farmacología clínica]. No se recomienda el uso de VIREAD® en pacientes pediátricos menores de 12 años o con un peso menor a 35 kg infectados por el VIH-1. Pacientes pediátricos de 12 años o más con hepatitis B crónica: En el estudio 115, 106 pacientes HBeAg negativos (9 %) y positivos (91%) de 12 a menos de 18 años de edad con infección por VHB crónica fueron aleatorizados a recibir tratamiento ciego con VIREAD® 300 mg (N = 52) o placebo (N = 54) durante 72 semanas. Al inicio del estudio, el valor medio de ADN de VHB fue de 8,1 log10 copias/ml y el valor medio de ALT fue de 101 U/l. De los 52 pacientes tratados con VIREAD®, 20 pacientes nunca habían recibido tratamiento previo con nucleós(t)idos y 32 pacientes habían recibido tratamiento previo con nucleós(t)idos. Treinta y uno de los 32 pacientes que habían recibido tratamiento previo con nucleós(t)idos también habían recibido tratamiento previo con lamivudina. En la semana 72, el 88 % (46/52) de los pacientes en el grupo de VIREAD® y un 0 % (0/54) de los pacientes en el grupo de placebo presentaron un nivel de ADN de VHB < 400 copias/ml (69 UI/ml). Entre los pacientes con valores ALT anormales al inicio, el 74 % (26/35) de los pacientes que recibieron VIREAD® presentó normalización de la ALT en la semana 72 frente al 31 % (13/42) de los pacientes en el grupo de placebo. Un paciente tratado con VIREAD® presentó pérdida de HBsAg sostenida y seroconversión a anti-HB durante las primeras 72 semanas de su participación en el estudio. VIREAD® no esta recomendado en los pacientes pediátricos menores de 12 años o con un peso inferior a 35 kg con hepatitis B crónica. Uso geriátrico: Los estudios clínicos de VIREAD® no incluyeron un número suficiente de pacientes de 65 años o más como para determinar si responden de forma diferente frente a los pacientes más jóvenes. En general, la selección de la dosis para los pacientes de edad avanzada debe ser cautelosa y se debe tener en cuenta una mayor frecuencia de disminución de las funciones cardíaca, renal o hepática, y las enfermedades concomitantes u otros tratamientos con fármacos. Pacientes con disfunción renal: Se recomienda modificar el intervalo de las dosis de VIREAD® en los pacientes con una depuración de creatinina estimada inferior a 50 ml/min y en los pacientes con enfermedad renal terminal que precisan diálisis [véase Dosificación, Farmacología clínica]. Estudios Clínicos: Eficacia clínica en pacientes con infección por VIH-1 Pacientes adultos sin tratamiento previo con antirretrovirales: Estudio 903: Se notifican los datos obtenidos hasta la semana 144 en el estudio 903, un estudio multicéntrico, aleatorizado y con control activo, en el que se comparó la administración de VIREAD® (300 mg una vez al día) en combinación con lamivudina y efavirenz, frente a estavudina (d4T), lamivudina y efavirenz, en 600 pacientes sin tratamiento previo con antirretrovirales. Los pacientes tenían una media de edad de 36 años (intervalo de 18 a 64 años), el 74 % eran varones, el 64 % era de raza blanca, y el 20 % era de raza negra. El recuento medio inicial de linfocitos CD4+ fue de 279 linfocitos/ mm3 (intervalo de 3 a 956), y la mediana del ARN del VIH-1 inicial en el plasma fue de 77.600 copias/ml (intervalo de 417 a 5.130.000). Los pacientes se estratificaron según el recuento inicial de linfocitos CD4+ y el ARN del VIH-1. Un 43 % de los pacientes tenía cargas virales iniciales > 100.000 copias/ml, y un 39 % tenía recuentos de linfocitos CD4+ < 200 linfocitos/ mm3. En la tabla 8 se presentan los resultados del tratamiento después de 48 y de 144 semanas.
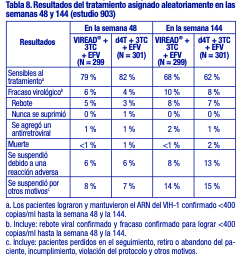
El logro de concentraciones del ARN del VIH-1 plasmático inferiores a 400 copias/ml en la semana 144 fue similar entre los dos grupos de tratamiento, en cuanto a la población estratificada según el valor inicial de concentración de ARN del VIH-1 ( > o ≤100 000 copias/ml) y recuento de linfocitos CD4+ ( < o ≥ 200 linfocitos/ mm3). Hasta la semana 144 de tratamiento, el 62 % y el 58 % de los pacientes en los grupos de VIREAD® y estavudina, respectivamente, lograron y mantuvieron el ARN del VIH-1 confirmado < 50 copias/ml. El aumento promedio del recuento de linfocitos CD4+ con respecto al valor inicial fue de 263 linfocitos/ mm3 en el grupo tratado con VIREAD®, y de 283 linfocitos/ mm3 en el grupo que recibió estavudina. Hasta la semana 144, 11 pacientes del grupo tratado con VIREAD® y 9 pacientes del grupo tratado con estavudina sufrieron una reacción nueva de Clase C según el código de los CDC. Estudio 934: Se notifican los datos obtenidos hasta la semana 144 en el estudio 934, un estudio multicéntrico, aleatorizado, abierto y con control activo, en el que se comparó la administración de emtricitabina + VIREAD® en combinación con efavirenz, frente a la asociación en dosis fijas de zidovudina y lamivudina en combinación con efavirenz, en 511 pacientes sin tratamiento previo con antirretrovirales. De la semana 96 a la 144 del estudio, los pacientes recibieron una asociación en dosis fijas de emtricitabina + tenofovir DF con efavirenz en lugar de emtricitabina + VIREAD® con efavirenz. Los pacientes tenían una media de edad de 38 años (intervalo de 18 a 80 años), el 86 % eran varones, el 59 % era de raza blanca, y el 23 % era de raza negra. El recuento medio inicial de linfocitos CD4+ fue de 245 linfocitos/ mm3 (intervalo de 2 a 1191), y la mediana del ARN del VIH1 inicial en el plasma fue de 5,01 log10 copias/ml (intervalo de 3,56 a 6,54). Los pacientes se estratificaron según el recuento inicial de linfocitos CD4+ ( < o ≥200 linfocitos/ mm3); el 41 % tenía recuentos de linfocitos CD4+ < 200 linfocitos/ mm3, y el 51 % tenía cargas virales iniciales > 100 000 copias/ml. En la tabla 9 se presentan los resultados del tratamiento después de 48 y de 144 semanas de tratamiento en los pacientes que no presentaron resistencia inicial al efavirenz.
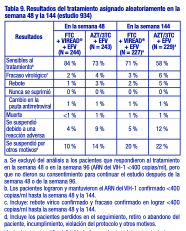
Hasta la semana 48, el 84 % de los pacientes del grupo tratado con emtricitabina + VIREAD® y el 73 % de los pacientes del grupo tratado con zidovudina y lamivudina lograron y mantuvieron el ARN del VIH-1 < 400 copias/ml (71 % y 58 %, respectivamente, hasta la semana 144). En este estudio abierto, la diferencia en la proporción de pacientes que lograron y mantuvieron el ARN del VIH-1 < 400 copias/ml después de 48 semanas de tratamiento se debe principalmente a un mayor número de suspensiones del tratamiento por reacciones adversas y otros motivos en el grupo tratado con zidovudina y lamivudina. Además, el 80 % de los pacientes del grupo tratado con emtricitabina + VIREAD® y el 70 % de los pacientes del grupo tratado con zidovudina y lamivudina lograron y mantuvieron el ARN del VIH-1 < 50 copias/ml hasta la semana 48 (64% y 56 %, respectivamente, hasta la semana 144). En la semana 48, el aumento medio con respecto a los valores iniciales del recuento de linfocitos CD4+ fue de 190 linfocitos/ mm3 en el grupo tratado con EMTRIVA® (emtricitabina) + VIREAD®, y de 158 linfocitos/ mm3 en el grupo que recibió zidovudina y lamivudina (312 y 271 linfocitos/ mm3, respectivamente, en la semana 144). A las 48 semanas, siete pacientes del grupo tratado con emtricitabina + VIREAD® y cinco pacientes del grupo tratado con zidovudina y lamivudina experimentaron una nueva reacción de clase C según el código de los CDC (10 y 6 pacientes, respectivamente, hasta la semana 144). Pacientes adultos con tratamiento previo: Estudio 907: El estudio 907 fue un estudio multicéntrico, doble ciego, controlado con placebo y de 24 semanas de duración con VIREAD® agregado a una pauta de fondo estable de antirretrovirales en 550 pacientes que habían recibido tratamiento previo con antirretrovirales. Después de 24 semanas de tratamiento del estudio con ciego, se ofreció abiertamente VIREAD® a todos los pacientes que continuaron el estudio durante un periodo adicional de 24 semanas. Los pacientes tuvieron un recuento medio inicial de linfocitos CD4+ de 427 linfocitos/ mm3 (intervalo entre 23 y 1385), una mediana del ARN del VIH-1 inicial en el plasma de 2340 (intervalo entre 50 y 75 000) copias/ml y una duración promedio del tratamiento de la infección por el VIH-1 previo de 5,4 años. La media de edad de los pacientes era de 42 años, el 85 % eran varones, el 69 % eran de raza blanca, el 17 % eran de raza negra, y el 12% eran de origen hispano. En la tabla 10 se resumen el porcentaje de pacientes con ARN del VIH-1 < 400 copias/ml y los resultados de los pacientes después de 48 semanas.
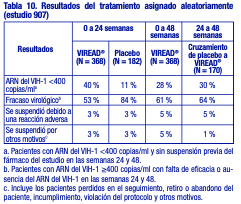
Tras 24 semanas de tratamiento, hubo una proporción mayor de pacientes en el grupo tratado con VIREAD® con ARN del VIH-1 < 50 copias/ml (19 %), en comparación con el grupo que recibió placebo (1 %). El cambio promedio en los recuentos absolutos de linfocitos CD4+ en la semana 24 fue de +11 linfocitos/ mm3 en el grupo que recibió VIREAD® y de -5 linfocitos/ mm3 en el grupo que recibió placebo. El cambio promedio en los recuentos absolutos de linfocitos CD4+ en la semana 48 fue de +4 linfocitos/ mm3 en el grupo tratado con VIREAD®. Hasta la semana 24, un paciente del grupo que recibió VIREAD® y ningún paciente del grupo que recibió placebo presentaron una reacción nueva de Clase C según el código de los CDC. Eficacia clínica en pacientes adultos con hepatitis B crónica: Hepatitis B crónica HBeAg negativa: El estudio 0102 fue un estudio de fase 3, aleatorizado, doble ciego, con control activo, de 300 mg de VIREAD® en comparación con 10 mg de HEPSERA® (dipivoxilo de adefovir), realizado en 375 pacientes HBeAg(anti-HBe+) con función hepática compensada, la mayoría de los cuales no había recibido nucleósidos previamente. La media de edad de los pacientes era de 44 años, el 77 % eran varones, el 25 % eran de raza asiática, el 65 % eran de raza blanca, el 17 % habían recibido tratamiento previo con interferón alfa, y el 18% habían recibido nucleósidos previamente (el 16 % había recibido tratamiento previo con lamivudina). Al inicio, los pacientes tenían una media de 7,8 en la puntuación necroinflamatoria de Knodell; la media del ADN de VHB plasmático era de 6,9 log10 copias/ml; y la media de la ALT sérica era de 140 U/l. Hepatitis B crónica HBeAg positiva: El estudio 0103 fue un estudio de fase 3, aleatorizado, doble ciego, con control activo, de 300 mg de VIREAD® en comparación con 10 mg de HEPSERA® (dipivoxilo de adefovir), realizado en 266 pacientes HBeAg+ con función hepática compensada que no habían recibido previamente nucleósidos. La media de edad de los pacientes era de 34 años, el 69 % eran varones, el 36 % eran de raza asiática, el 52 % eran de raza blanca, el 16 % habían recibido tratamiento previo con interferón alfa, y < 5 % habían recibido nucleósidos previamente. Al inicio, los pacientes tenían una media de 8,4 en la puntuación necroinflamatoria de Knodell; la media del ADN de VHB plasmático era de 8,7 log10 copias/ml; y la media de la ALT sérica era de 147 U/l. El análisis de los datos primarios se realizó después de que todos los pacientes alcanzaran las 48 semanas de tratamiento; los resultados se resumen a continuación. El criterio de valoración primario de eficacia en ambos estudios fue la respuesta completa al tratamiento, definida como un nivel de ADN de VHB < 400 copias/ml (69 UI/ml) y una mejora de al menos 2 puntos en la puntuación necroinflamatoria de Knodell, sin empeoramiento en la fibrosis de Knodell en la semana 48 (tabla 11).
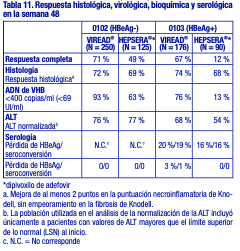
Tratamiento durante más de 48 semanas: En los estudios 0102 (HBeAg negativo) y 0103 (HBeAg positivo), los pacientes fueron elegibles para pasar a recibir VIREAD® abiertamente sin interrupción del tratamiento después de completar el tratamiento con doble ciego (389 y 196 pacientes que fueron inicialmente aleatorizados a VIREAD® y HEPSERA®,respectivamente). En el estudio 0102, 266 de los 347 pacientes que ingresaron en el período sin enmascaramiento (77 %) continuaron en el estudio hasta la semana 384. Entre los pacientes aleatorizados a VIREAD® seguido del tratamiento abierto con VIREAD®, el 73% tenía un nivel de ADN de VHB < 400 copias/ml (69 UI/ml) y el 63% presentó normalización de la ALT en la semana 384. En el grupo de los pacientes aleatorizados a HEPSERA® (dipivoxilo de adefovir) seguido del tratamiento abierto con VIREAD®, el 80% tenía un nivel de ADN de VHB < 400 copias/ml (69 UI/ml) y el 76 % presentó normalización de la ALT hasta la semana 384. En la semana 384, tanto la pérdida como la seroconversión de HBsAg fueron aproximadamente del 1% en ambos grupos de tratamiento. En el estudio 0103, 146 de los 238 pacientes que ingresaron en el período sin enmascaramiento (61%) continuaron en el estudio hasta la semana 384. Entre los pacientes aleatorizados a VIREAD®, el 49% tenía un nivel de ADN de VHB < 400 copias/ml (69 UI/ml), el 42% presentó normalización de la ALT y el 20% tuvo pérdida de HBeAg (13% de seroconversión a anticuerpos anti-HBe) hasta la semana 384. Entre los pacientes aleatorizados a HEPSERA® (dipivoxilo de adefovir) seguido por un tratamiento abierto con VIREAD®, el 56% tenía un nivel de ADN de VHB < 400 copias/ml (69 UI/ml), el 50% presentó normalización de la ALT y el 28% tuvo pérdida de HBeAg (19% de seroconversión a anticuerpos anti-HBe) hasta la semana 384. La pérdida y la seroconversión de HBsAg hasta la semana 384 fueron del 11% y del 8% respectivamente en en los pacientes aleatorizados inicialmente a VIREAD® y del 12% y del 10% respectivamente en los pacientes aleatorizados inicialmente a HEPSERA® (dipivoxilo de adefovir). De los 641 pacientes que fueron inicialmente aleatorizados y tratados en los dos estudios, estaban disponibles para análisis los datos de biopsias hepáticas de 328 pacientes que recibieron tratamiento abierto continuo con VIREAD® en monoterapia al inicio, la semana 48 y la semana 240. No se identificaron diferencias aparentes entre el subgrupo de pacientes que tuvieron datos de biopsias hepáticas en la semana 240 y aquellos pacientes que siguieron recibiendo VIREAD® abierto sin los datos de la biopsia que se esperaba que afectase